Rebetol 200mg Ribavirin Anvendelse, bivirkninger og dosering. Pris i onlineapotek. Generisk medicin uden recept.
Hvad er Rebetol 200mg, og hvordan bruges det?
Rebetol er en receptpligtig medicin, der bruges til at behandle symptomerne på kronisk hepatitis C. Rebetol 200mg kan bruges alene eller sammen med anden medicin.
Rebetol tilhører en klasse af lægemidler kaldet hepatitis B/hepatitis C-midler; RSV-agenter.
Det vides ikke, om Rebetol 200mg er sikkert og effektivt til børn under 3 år.
Hvad er de mulige bivirkninger af Rebetol?
Rebetol kan forårsage alvorlige bivirkninger, herunder:
- nældefeber,
- åndedrætsbesvær,
- hævelse af dit ansigt, læber, tunge eller hals,
- synsproblemer
- stærke smerter i din øvre mave breder sig til ryggen,
- kvalme,
- opkastning,
- diarré,
- ny eller forværret hoste,
- feber,
- stikkende brystsmerter,
- hvæsen,
- stakåndet,
- svær depression,
- tanker om selvskade,
- tanker om at såre andre,
- bleg eller gulnet hud,
- mørk farvet urin,
- forvirring,
- svaghed,
- kuldegysninger,
- influenzalignende symptomer,
- hævede tandkød,
- sår i munden,
- hudsår,
- let blå mærker,
- usædvanlig blødning, og
- svimmelhed
- Få lægehjælp med det samme, hvis du har nogle af symptomerne nævnt ovenfor.
- De mest almindelige bivirkninger af Rebetol omfatter:
- kvalme,
- opkastning,
- mistet appetiten,
- feber,
- kuldegysninger,
- ryster,
- lavt antal blodlegemer,
- anæmi,
- svaghed,
- træthed,
- hovedpine,
- muskelsmerter,
- humørsvingninger,
- angst, og
- irritabilitet
Fortæl det til lægen, hvis du har en bivirkning, der generer dig, eller som ikke forsvinder.
Disse er ikke alle de mulige bivirkninger af Rebetol. Spørg din læge eller apotek for mere information.
Ring til din læge for lægehjælp om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
ADVARSEL
RISIKO FOR ALVORLIGE SYDELSER OG RIBAVIRIN-ASSOCIEREDE VIRKNINGER
- REBETOL monoterapi er ikke effektiv til behandling af kronisk hepatitis C-virusinfektion og bør ikke anvendes alene til denne indikation [se ADVARSLER OG FORHOLDSREGLER].
- Den primære toksicitet af ribavirin er hæmolytisk anæmi. Den anæmi, der er forbundet med behandling med REBETOL 200 mg, kan resultere i forværring af hjertesygdom, som har ført til dødelige og ikke-dødelige myokardieinfarkter. Patienter med en historie med signifikant eller ustabil hjertesygdom bør ikke behandles med REBETOL [se DOSERING OG ADMINISTRATION, ADVARSLER OG FORHOLDSREGLER, og BIVIRKNINGER].
- Signifikante teratogene og embryocidale virkninger er blevet påvist hos alle dyrearter, der er udsat for ribavirin. Derudover har ribavirin en halveringstid for flere doser på 12 dage, og det kan derfor forblive i ikke-plasma-kompartmenter i så længe som 6 måneder. Derfor er behandling med REBETOL kontraindiceret hos kvinder, der er gravide, og hos de mandlige partnere til kvinder, der er gravide. Der skal udvises ekstrem forsigtighed for at undgå graviditet under behandlingen og i 6 måneder efter afslutning af behandlingen hos både kvindelige patienter og hos kvindelige partnere til mandlige patienter, som tager REBETOL-behandling. Mindst to pålidelige former for effektiv prævention skal anvendes under behandlingen og i den 6-måneders opfølgningsperiode efter behandling [se KONTRAINDIKATIONER, ADVARSLER OG FORHOLDSREGLER, Brug i specifikke populationer, ikke-klinisk toksikologi og PATIENTINFORMATION].
BESKRIVELSE
REBETOL (ribavirin), er en syntetisk nukleosidanalog (purinanalog). Det kemiske navn på ribavirin er 1-β-D-ribofuranosyl-1H-1,2,4-triazol-3-carboxamid og har følgende strukturformel (se figur 1).
Figur 1: Strukturformel
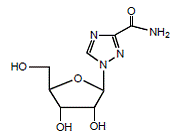
Ribavirin er et hvidt, krystallinsk pulver. Det er frit opløseligt i vand og let opløseligt i vandfri alkohol. Den empiriske formel er C8H12N4O5 og molekylvægten er 244,21.
REBETOL kapsler består af et hvidt pulver i en hvid, uigennemsigtig gelatinekapsel. Hver kapsel indeholder 200 mg ribavirin og de inaktive ingredienser mikrokrystallinsk cellulose, lactosemonohydrat, croscarmellosenatrium og magnesiumstearat. Kapselskallen består af gelatine, natriumlaurylsulfat, siliciumdioxid og titaniumdioxid. Kapslen er trykt med spiseligt blåt farmaceutisk blæk, som er lavet af shellak, vandfri ethylalkohol, isopropylalkohol, n-butylalkohol, propylenglycol, ammoniumhydroxid og FD&C Blue #2 aluminium lake.
REBETOL 200 mg oral opløsning er en klar, farveløs til bleg eller lysegul væske med tyggegummi-smag. Hver milliliter af opløsningen indeholder 40 mg ribavirin og de inaktive ingredienser saccharose, glycerin, sorbitol, propylenglycol, natriumcitrat, citronsyre, natriumbenzoat, naturlig og kunstig smag til tyggegummi #15864 og vand.
INDIKATIONER
Kronisk hepatitis C (CHC)
REBETOL® (ribavirin) i kombination med interferon alfa-2b (pegyleret og ikke-pegyleret) er indiceret til behandling af kronisk hepatitis C (CHC) hos patienter 3 år og ældre med kompenseret leversygdom [se ADVARSLER OG FORHOLDSREGLER , og Brug i specifikke populationer ].
Følgende punkter bør overvejes, når REBETOL kombinationsbehandling med PegIntron® eller INTRON A® påbegyndes:
- Disse indikationer er baseret på opnåelse af upåviselig HCV-RNA efter behandling i 24 eller 48 uger og opretholdelse af et vedvarende virologisk respons (SVR) 24 uger efter den sidste dosis.
- Kombinationsterapi med REBETOL/PegIntron foretrækkes frem for REBETOL/INTRON A, da denne kombination giver væsentligt bedre responsrater [se Kliniske Studier ].
- Patienter med følgende karakteristika er mindre tilbøjelige til at få gavn af genbehandling efter at have svigtet et behandlingsforløb: tidligere non-respons, tidligere pegyleret interferonbehandling, signifikant brodannende fibrose eller cirrhose og genotype 1-infektion [se Kliniske Studier ].
- Ingen sikkerheds- og effektdata er tilgængelige for behandling på mere end et år.
DOSERING OG ADMINISTRATION
REBETOL kapsler må under ingen omstændigheder åbnes, knuses eller knækkes. REBETOL 200mg bør tages sammen med mad [se KLINISK FARMAKOLOGI ]. REBETOL bør ikke anvendes til patienter med kreatininclearance mindre end 50 ml/min.
REBETOL/PegIntron kombinationsterapi
Voksne patienter
Den anbefalede dosis af PegIntron er 1,5 mcg/kg/uge subkutant i kombination med 800 til 1400 mg REBETOL 200 mg kapsler oralt baseret på patientens kropsvægt (se tabel 1). Mængden af PegIntron, der skal injiceres, afhænger af styrken af PegIntron og patientens kropsvægt. Se mærkningen for PegIntron for yderligere information om dosering.
Behandlingsvarighed – Interferon alfa-naive patienter
Behandlingsvarigheden for patienter med genotype 1 er 48 uger. Seponering af behandlingen bør overvejes hos patienter, som ikke opnår mindst 2 log10 fald eller tab af HCV-RNA efter 12 uger, eller hvis HCV-RNA forbliver påviselig efter 24 ugers behandling. Patienter med genotype 2 og 3 bør behandles i 24 uger.
Behandlingsvarighed – Genbehandling med PegIntron/REBETOL af tidligere behandlingssvigt
Behandlingsvarigheden for patienter, der tidligere har svigtet behandlingen, er 48 uger, uanset HCV-genotype. Genbehandlede patienter, som ikke opnår upåviselig HCV-RNA i uge 12 af behandlingen, eller hvis HCV-RNA forbliver påviselig efter 24 ugers behandling, er højst usandsynligt at opnå SVR, og seponering af behandlingen bør overvejes [se Kliniske Studier ].
Pædiatriske patienter
Dosering til pædiatriske patienter bestemmes af kropsoverfladeareal for PegIntron og af kropsvægt for REBETOL. Den anbefalede dosis af PegIntron er 60 mcg/m²/uge subkutant i kombination med 15 mg/kg/dag af REBETOL 200 mg oralt i to opdelte doser (se tabel 2) til pædiatriske patienter i alderen 3-17 år. Patienter, der når deres 18-års fødselsdag, mens de får PegIntron/REBETOL 200 mg, bør forblive på det pædiatriske doseringsregime. Behandlingsvarigheden for patienter med genotype 1 er 48 uger. Patienter med genotype 2 og 3 bør behandles i 24 uger.
REBETOL/INTRON En kombinationsterapi
Voksne
Behandlingsvarighed – Interferon alfa-naive patienter
Den anbefalede dosis af INTRON A er 3 millioner IE tre gange ugentligt subkutant. Den anbefalede dosis af REBETOL kapsler afhænger af patientens kropsvægt (se tabel 3). Den anbefalede behandlingsvarighed for patienter, der tidligere ikke er behandlet med interferon, er 24 til 48 uger. Behandlingsvarigheden bør individualiseres til patienten afhængigt af baseline sygdomskarakteristika, respons på terapi og kurens tolerabilitet [se INDIKATIONER OG BRUG , BIVIRKNINGER , og Kliniske Studier ]. Efter 24 ugers behandling bør virologisk respons vurderes. Afbrydelse af behandlingen bør overvejes hos enhver patient, som ikke har opnået et HCV-RNA under analysens detektionsgrænse inden for 24 uger. Der er ingen sikkerheds- og effektdata for behandling i mere end 48 uger i den tidligere ubehandlede patientpopulation.
Behandlingsvarighed – Genbehandling med INTRON A/REBETOL hos tilbagefaldspatienter
Hos patienter, som får tilbagefald efter ikke-pegyleret interferon monoterapi, er den anbefalede behandlingsvarighed 24 uger.
Pædiatri
Den anbefalede dosis af REBETOL er 15 mg/kg per dag oralt (delt dosis AM og PM). Se tabel 2 for pædiatrisk dosering af REBETOL i kombination med INTRON A. INTRON A til injektion efter kropsvægt på 25 kg til 61 kg er 3 millioner IE/m² tre gange ugentligt subkutant. Se doseringstabel for voksne for mere end 61 kg legemsvægt.
Den anbefalede behandlingsvarighed er 48 uger for pædiatriske patienter med genotype 1. Efter 24 ugers behandling bør virologisk respons vurderes. Afbrydelse af behandlingen bør overvejes hos enhver patient, som ikke har opnået et HCV-RNA under analysens detektionsgrænse på dette tidspunkt. Den anbefalede behandlingsvarighed til pædiatriske patienter med genotype 2/3 er 24 uger.
Laboratorieprøver
Følgende laboratorietest anbefales til alle patienter, der behandles med REBETOL 200 mg, før behandlingen påbegyndes og derefter periodisk derefter.
Standard hæmatologiske tests - inklusive hæmoglobin (forbehandling, uge 2 og uge 4 af behandlingen, og som klinisk passende [se ADVARSLER OG FORHOLDSREGLER ], fuldstændigt og differentielt antal hvide blodlegemer og blodpladetal.
- Blodkemi - leverfunktionstest og TSH.
- Graviditet - inklusive månedlig overvågning for kvinder i den fødedygtige alder.
- EKG [se ADVARSLER OG FORHOLDSREGLER ].
Dosisændringer
Hvis der udvikles alvorlige bivirkninger eller laboratorieabnormiteter under kombinationsbehandling med REBETOL/INTRON A eller REBETOL/PegIntron, skal dosis ændres eller afbrydes, indtil bivirkningen aftager eller falder i sværhedsgrad [se ADVARSLER OG FORHOLDSREGLER ]. Hvis intolerancen fortsætter efter dosisjustering, bør kombinationsbehandlingen seponeres. Dosisreduktion af PegIntron hos voksne patienter på REBETOL/PegIntron kombinationsbehandling opnås i en to-trins proces fra den oprindelige startdosis på 1,5 mcg/kg/uge til 1 mcg/kg/uge og derefter til 0,5 mcg/kg/uge , hvis det er nødvendigt. Se mærkningen for PegIntron for yderligere information om dosisreduktion af PegIntron.
I studie 2 med kombinationsterapi for voksne forekom dosisreduktioner hos 42 % af forsøgspersonerne, der fik PegIntron 1,5 mcg/kg og REBETOL 800 mg dagligt, inklusive 57 % af de forsøgspersoner, der vejede 60 kg eller derunder. I undersøgelse 4 fik 16 % af forsøgspersonerne en dosisreduktion af PegIntron til 1 mcg/kg i kombination med REBETOL, mens yderligere 4 % krævede den anden dosisreduktion af PegIntron til 0,5 mcg/kg på grund af bivirkninger [se BIVIRKNINGER ].
Dosisreduktion hos pædiatriske patienter opnås ved at ændre den anbefalede PegIntron-dosis i en to-trins proces fra den oprindelige startdosis på 60 mcg/m²/uge til 40 mcg/m²/uge, derefter til 20 mcg/m²/uge, hvis nødvendigt (se tabel 4). I det pædiatriske kombinationsterapiforsøg forekom dosisreduktioner hos 25 % af forsøgspersonerne, der fik PegIntron 60 mcg/m² ugentligt og REBETOL 15 mg/kg dagligt. Dosisreduktion hos pædiatriske patienter opnås ved at ændre den anbefalede REBETOL-dosis fra den oprindelige startdosis på 15 mg/kg dagligt i en to-trins proces til 12 mg/kg/dag, derefter til 8 mg/kg/dag, hvis det er nødvendigt ( se tabel 4).
REBETOL 200mg bør ikke anvendes til patienter med kreatininclearance mindre end 50 ml/min. Patienter med nedsat nyrefunktion og personer over 50 år bør overvåges nøje med hensyn til udvikling af anæmi [se ADVARSLER OG FORHOLDSREGLER , Brug i specifikke populationer , og KLINISK FARMAKOLOGI ].
REBETOL 200 mg bør administreres med forsigtighed til patienter med allerede eksisterende hjertesygdom. Patienter bør vurderes før påbegyndelse af behandlingen og bør overvåges passende under behandlingen. Hvis der er nogen forværring af kardiovaskulær status, skal behandlingen stoppes [se ADVARSLER OG FORHOLDSREGLER ].
For patienter med en anamnese med stabil kardiovaskulær sygdom er en permanent dosisreduktion påkrævet, hvis hæmoglobinet falder med mere end eller lig med 2 g/dL i løbet af en 4-ugers periode. Hvis hæmoglobinet forbliver mindre end 12 g/dL efter 4 uger på en reduceret dosis, bør patienten desuden afbryde kombinationsbehandlingen for disse hjertepatienter.
Det anbefales, at en patient, hvis hæmoglobinniveau falder til under 10 g/dL, får sin REBETOL-dosis ændret eller seponeret i henhold til tabel 4 [se ADVARSLER OG FORHOLDSREGLER ].
Se mærkningen for INTRON A eller PegIntron for yderligere information om, hvordan man reducerer en INTRON A- eller PegIntron-dosis.
Seponering af dosering
Voksne
Hos HCV genotype 1, interferon-alfa-naive patienter, der får PegIntron i kombination med ribavirin, anbefales det at seponere behandlingen, hvis der ikke er mindst 2 log10 fald eller tab af HCV-RNA efter 12 ugers behandling, eller hvis HCV-RNA niveauer forbliver påviselige efter 24 ugers behandling. Uanset genotype er det højst usandsynligt, at tidligere behandlede patienter, som har påviselig HCV-RNA i uge 12 eller 24, opnår SVR, og seponering af behandlingen bør overvejes.
Pædiatri (3-17 år)
Det anbefales, at patienter, der får PegIntron/REBETOL-kombination (undtagen HCV genotype 2 og 3), seponeres fra behandlingen efter 12 uger, hvis deres behandling Uge 12 HCV-RNA faldt mindre end 2 log10 sammenlignet med en forbehandling eller efter 24 uger, hvis de har påviselig HCV-RNA ved behandling uge 24.
HVORDAN LEVERET
Doseringsformer og styrker
REBETOL 200mg Kapsler 200 mg
REBETOL 200 mg oral opløsning 40 mg pr. ml
Opbevaring og håndtering
REBETOL 200 mg kapsler er hvide, uigennemsigtige kapsler med REBETOL, 200 mg, og Schering Corporation-logoet påtrykt kapselskallen; kapslerne er pakket i en flaske indeholdende 56 kapsler ( NDC 0085-1351-05), 70 kapsler ( NDC 0085-1385-07) og 84 kapsler ( NDC 0085-1194-03).
REBETOL 200mg oral opløsning 40 mg ml er en klar, farveløs til bleg eller lysegul væske med tyggegummi-smag, og den er pakket i 4-oz ravfarvede glasflasker (100 ml/flaske) med børnesikre lukninger ( NDC 0085-1318-01).
Flasken med REBETOL kapsler skal opbevares ved 25°C (77°F); udflugter tilladt til 15-30°C (59-86°F) [se USP kontrolleret rumtemperatur ].
REBETOL 200mg oral opløsning skal opbevares mellem 2-8°C (36-46°F) eller ved 25°C (77°F); udflugter tilladt til 15-30°C (59-86°F) [se USP kontrolleret rumtemperatur ].
REBETOL 200mg oral opløsning fremstillet til: Merck Sharp & Dohme Corp., et datterselskab af Whitehouse Station, NJ 08889, USA. Fremstillet af: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Canada REBETOL Kapsler fremstillet af: Merck Sharp & Dohme Corp., et datterselskab af Whitehouse Station, NJ 08889, USA. Revideret: december 2014.
BIVIRKNINGER
Kliniske forsøg med REBETOL i kombination med PegIntron eller INTRON A er blevet udført i over 7800 forsøgspersoner fra 3 til 76 år.
Den primære toksicitet af ribavirin er hæmolytisk anæmi. Reduktioner i hæmoglobinniveauer forekom inden for de første 1 til 2 uger af oral behandling. Hjerte- og lungereaktioner forbundet med anæmi forekom hos ca. 10 % af patienterne [se ADVARSLER OG FORHOLDSREGLER ].
Mere end 96 % af alle forsøgspersoner i kliniske forsøg oplevede en eller flere bivirkninger. De mest almindeligt rapporterede bivirkninger hos voksne forsøgspersoner, der fik PegIntron eller INTRON A i kombination med REBETOL 200 mg, var inflammation/reaktion på injektionsstedet, træthed/asteni, hovedpine, stivhed, feber, kvalme, myalgi og angst/emotionel labilitet/irritabilitet. De mest almindelige bivirkninger hos pædiatriske forsøgspersoner i alderen 3 og ældre, der fik REBETOL 200 mg i kombination med PegIntron eller INTRON A, var pyreksi, hovedpine, neutropeni, træthed, anoreksi, erytem på injektionsstedet og opkastning.
Bivirkningssektionen henviser til følgende kliniske forsøg:
- REBETOL/PegIntron Kombinationsterapi forsøg:
- Klinisk undersøgelse 1 – evalueret PegIntron monoterapi (ikke yderligere beskrevet på denne etiket; se mærkning af PegIntron for information om dette forsøg).
- Studie 2 – evalueret REBETOL 800 mg/dag flad dosis i kombination med 1,5 mcg/kg/uge PegIntron eller med INTRON A.
- Studie 3 – evalueret PegIntron/vægtbaseret REBETOL 200mg i kombination med PegIntron/fladdosis REBETOL 200mg regime.
- Undersøgelse 4 – sammenlignede to doser PegIntron (1,5 mcg/kg/uge og 1 mcg/kg/uge) i kombination med REBETOL og en tredje behandlingsgruppe, der fik Pegasys® (180 mcg/uge)/Copegus® (1000-1200 mg/dag) ).
- Undersøgelse 5 – evaluerede PegIntron (1,5 mcg/kg/uge) i kombination med vægtbaseret REBETOL hos forsøgspersoner med tidligere behandlingssvigt.
- PegIntron/REBETOL 200mg kombinationsterapi til pædiatriske patienter
- REBETOL/INTRON A kombinationsterapiforsøg for voksne og pædiatri
Alvorlige bivirkninger er forekommet hos ca. 12 % af forsøgspersonerne i kliniske forsøg med PegIntron med eller uden REBETOL [se KASSET ADVARSEL , ADVARSLER OG FORHOLDSREGLER ]. De mest almindelige alvorlige hændelser, der opstod hos forsøgspersoner behandlet med PegIntron og REBETOL 200 mg, var depression og selvmordstanker [se ADVARSLER OG FORHOLDSREGLER ], der hver forekommer med en frekvens på mindre end 1 %. Selvmordstanker eller selvmordsforsøg forekom hyppigere blandt pædiatriske patienter, primært teenagere, sammenlignet med voksne patienter (2,4 % versus 1 %) under behandling og opfølgning uden for behandlingen [se ADVARSLER OG FORHOLDSREGLER ]. Den mest almindelige fatale reaktion, der opstod hos forsøgspersoner behandlet med PegIntron og REBETOL 200 mg, var hjertestop, selvmordstanker og selvmordsforsøg [se ADVARSLER OG FORHOLDSREGLER ], alle forekommer hos mindre end 1 % af forsøgspersonerne.
Fordi kliniske forsøg udføres under vidt forskellige forhold, kan antallet af bivirkninger observeret i de kliniske forsøg med et lægemiddel ikke direkte sammenlignes med frekvenserne i de kliniske forsøg med et andet lægemiddel og afspejler muligvis ikke de hyppigheder, der observeres i klinisk praksis.
Erfaring med kliniske forsøg – REBETOL/PegIntron kombinationsterapi
Voksne emner
Bivirkninger, der opstod i det kliniske forsøg med en forekomst på mere end 5 %, er angivet efter behandlingsgruppe fra REBETOL/PegIntron-kombinationsterapien (undersøgelse 2) i tabel 5.
Tabel 6 opsummerer de behandlingsrelaterede bivirkninger i undersøgelse 4, der forekom med en hyppighed på mere end eller lig med 10 %.
Hyppigheden af alvorlige bivirkninger var sammenlignelig i alle forsøg. I undersøgelse 3 var der en lignende forekomst af alvorlige bivirkninger rapporteret for den vægtbaserede REBETOL-gruppe (12 %) og for fladdosis-REBETOL-regimet. I undersøgelse 2 var forekomsten af alvorlige bivirkninger 17 % i PegIntron/REBETOL-grupperne sammenlignet med 14 % i INTRON A/REBETOL-gruppen.
mange, men ikke alle tilfælde, forsvandt bivirkningerne efter dosisreduktion eller seponering af behandlingen. Nogle forsøgspersoner oplevede igangværende eller nye alvorlige bivirkninger i løbet af den 6-måneders opfølgningsperiode. I undersøgelse 2 fortsatte mange forsøgspersoner med at opleve bivirkninger flere måneder efter seponering af behandlingen. Ved udgangen af den 6-måneders opfølgningsperiode var forekomsten af igangværende bivirkninger efter kropsklasse i PegIntron 1,5/REBETOL 200 mg-gruppen 33 % (psykiatrisk), 20 % (muskuloskeletal) og 10 % (for endokrine og for GI). Hos cirka 10 til 15 % af forsøgspersonerne var vægttab, træthed og hovedpine ikke forsvundet.
Der har været 31 forsøgspersoners dødsfald, der opstod under behandling eller under opfølgning i disse kliniske forsøg. I undersøgelse 1 var der 1 selvmord hos en forsøgsperson, der fik PegIntron monoterapi og 2 dødsfald blandt forsøgspersoner, der fik INTRON A monoterapi (1 mord/selvmord og 1 pludselig død). I undersøgelse 2 var der 1 selvmord hos en forsøgsperson, der fik PegIntron/REBETOL 200 mg kombinationsbehandling; og 1 forsøgsperson død i INTRON A/REBETOL 200mg-gruppen (motorkøretøjsulykke). I undersøgelse 3 var der 14 dødsfald, hvoraf 2 var sandsynlige selvmord og 1 var et uforklarligt dødsfald hos en person med en relevant sygehistorie med depression. I undersøgelse 4 var der 12 dødsfald, hvoraf 6 forekom hos forsøgspersoner, der fik PegIntron/REBETOL kombinationsbehandling, 5 i PegIntron 1,5 mcg/REBETOL-armen (N=1019) og 1 i PegIntron 1 mcg/REBETOL-armen (N= 1016), og 6 af dem forekom hos forsøgspersoner, der fik Pegasys/Copegus (N=1035); der var 3 selvmord, der fandt sted under opfølgningsperioden uden for behandlingen hos forsøgspersoner, som fik PegIntron (1,5 mcg/kg)/REBETOL 200 mg kombinationsbehandling.
undersøgelse 1 og 2 ophørte 10 til 14 % af forsøgspersonerne, der fik PegIntron, alene eller i kombination med REBETOL 200 mg, behandlingen sammenlignet med 6 % behandlet med INTRON A alene og 13 % behandlet med INTRON A i kombination med REBETOL. Tilsvarende i undersøgelse 3 afbrød 15 % af forsøgspersonerne, der fik PegIntron i kombination med vægtbaseret REBETOL, og 14 % af forsøgspersonerne, der fik PegIntron og en flad dosis REBETOL 200 mg, behandlingen på grund af en bivirkning. De mest almindelige årsager til seponering af behandlingen var relateret til kendte interferoneffekter af psykiatriske, systemiske (f.eks. træthed, hovedpine) eller gastrointestinale bivirkninger. I undersøgelse 4 afbrød 13 % af forsøgspersonerne i PegIntron 1,5 mcg/REBETOL 200 mg-armen, 10 % i PegIntron 1 mcg/REBETOL 200 mg-armen og 13 % i Pegasys 180 mcg/Copegus-armen på grund af bivirkninger.
undersøgelse 2 forekom dosisreduktioner på grund af bivirkninger hos 42 % af forsøgspersonerne, der fik PegIntron (1,5 mcg/kg)/REBETOL 200 mg og hos 34 % af dem, der fik INTRON A/REBETOL. Størstedelen af forsøgspersoner (57 %), der vejede 60 kg eller derunder, som fik PegIntron (1,5 mcg/kg)/REBETOL, krævede dosisreduktion. Reduktion af interferon var dosisrelateret (PegIntron 1,5 mcg/kg større end PegIntron 0,5 mcg/kg eller INTRON A), henholdsvis 40%, 27%, 28%. Dosisreduktion for REBETOL 200 mg var ens på tværs af alle tre grupper, 33 til 35 %. De mest almindelige årsager til dosisændringer var neutropeni (18 %) eller anæmi (9 %) (se Laboratorieværdier ). Andre almindelige årsager omfattede depression, træthed, kvalme og trombocytopeni. I undersøgelse 3 forekom dosisændringer på grund af bivirkninger hyppigere med vægtbaseret dosering (WBD) sammenlignet med flad dosering (henholdsvis 29 % og 23 %). I undersøgelse 4 fik 16 % af forsøgspersonerne en dosisreduktion af PegIntron til 1 mcg/kg i kombination med REBETOL 200 mg, mens yderligere 4 % krævede den anden dosisreduktion af PegIntron til 0,5 mcg/kg på grund af bivirkninger sammenlignet med 15 %. af forsøgspersoner i Pegasys/Copegus-armen, som krævede en dosisreduktion til 135 mcg/uge med Pegasys, med yderligere 7 % i Pegasys/Copegus-armen, der krævede anden dosisreduktion til 90 mcg/uge med Pegasys.
PegIntron/REBETOL 200 mg kombinationsforsøgene var de mest almindelige bivirkninger psykiatriske, som forekom blandt 77 % af forsøgspersonerne i undersøgelse 2 og 68 % til 69 % af forsøgspersonerne i undersøgelse 3. Disse psykiatriske bivirkninger omfattede oftest depression, irritabilitet og søvnløshed, hver rapporteret af ca. 30 % til 40 % af forsøgspersonerne i alle behandlingsgrupper. Selvmordsadfærd (ideer, forsøg og selvmord) forekom hos 2 % af alle forsøgspersoner under behandling eller under opfølgning efter behandlingsophør [se ADVARSLER OG FORHOLDSREGLER ]. I undersøgelse 4 forekom psykiatriske bivirkninger hos 58 % af forsøgspersonerne i PegIntron 1,5 mcg/REBETOL 200 mg-armen, 55 % af forsøgspersonerne i PegIntron 1 mcg/REBETOL 200 mg-armen og 57 % af forsøgspersonerne i Pegasys-armen/180 mcg. .
PegIntron inducerede træthed eller hovedpine hos ca. to tredjedele af forsøgspersonerne, med feber eller stivhed hos ca. halvdelen af forsøgspersonerne. Sværhedsgraden af nogle af disse systemiske symptomer (f.eks. feber og hovedpine) havde en tendens til at falde, efterhånden som behandlingen fortsatte. I undersøgelse 1 og 2 forekom inflammation og reaktion på applikationsstedet (f.eks. blå mærker, kløe og irritation) med cirka dobbelt så stor forekomst med PegIntron-behandlinger (hos op til 75 % af forsøgspersonerne) sammenlignet med INTRON A. Smerter på injektionsstedet var imidlertid sjældent (2 til 3%) i alle grupper. I undersøgelse 3 var der en samlet forekomst på 23 % til 24 % for reaktioner på injektionsstedet eller betændelse.
Forsøgspersoner, der fik REBETOL/PegIntron som genbehandling efter at have svigtet et tidligere interferonkombinationsregime, rapporterede bivirkninger svarende til dem, der tidligere var forbundet med dette regime under kliniske forsøg med behandlingsnaive forsøgspersoner.
Pædiatriske emner
Generelt svarede bivirkningsprofilen i den pædiatriske population til den, der blev observeret hos voksne. I det pædiatriske forsøg var de mest udbredte bivirkninger hos alle forsøgspersoner pyreksi (80%), hovedpine (62%), neutropeni (33%), træthed (30%), anoreksi (29%), erytem på injektionsstedet (29). %) og opkastning (27 %). Størstedelen af de rapporterede bivirkninger i forsøget var milde eller moderate i sværhedsgrad. Alvorlige bivirkninger blev rapporteret hos 7 % (8/107) af alle forsøgspersoner og omfattede smerter på injektionsstedet (1 %), smerter i ekstremiteter (1 %), hovedpine (1 %), neutropeni (1 %) og pyreksi (4 %). %). Vigtige bivirkninger, der opstod i denne patientpopulation, var nervøsitet (7%; 7/107), aggression (3%; 3/107), vrede (2%; 2/107) og depression (1%; 1/107) . Fem forsøgspersoner modtog levothyroxinbehandling, tre med klinisk hypothyroidisme og to med asymptomatiske TSH-stigninger. Vægt- og højdeforøgelse hos pædiatriske forsøgspersoner, der blev behandlet med PegIntron plus REBETOL, var bagefter det, der blev forudsagt af normative populationsdata for hele behandlingens længde. Svært hæmmet væksthastighed (mindre end 3. percentil) blev observeret hos 70 % af forsøgspersonerne under behandling.
Dosisændringer af PegIntron og/eller ribavirin var påkrævet hos 25 % af forsøgspersonerne på grund af behandlingsrelaterede bivirkninger, oftest for anæmi, neutropeni og vægttab. To forsøgspersoner (2 %; 2/107) ophørte med behandlingen som følge af en bivirkning.
Bivirkninger, der opstod med en forekomst på mere end eller lig med 10 % hos de pædiatriske forsøgspersoner, er angivet i tabel 7.
Fireoghalvfems ud af 107 forsøgspersoner blev tilmeldt et 5-årigt langsigtet opfølgningsforsøg. De langsigtede virkninger på væksten var mindre hos de forsøgspersoner, der blev behandlet i 24 uger, end dem, der blev behandlet i 48 uger. Fireogtyve procent af forsøgspersonerne (11/46) behandlet i 24 uger og 40% af forsøgspersonerne (19/48) behandlet i 48 uger havde et fald på > 15 percentil højde for alder fra forbehandling til slutningen af 5 år langtidsopfølgning sammenlignet med før-behandlings baseline percentiler. Elleve procent af forsøgspersonerne (5/46) behandlet i 24 uger og 13% af forsøgspersonerne (6/48) behandlet i 48 uger blev observeret at have et fald fra før-behandlingens baseline på > 30 højde-for-alder percentiler til slutningen af den 5-årige langtidsopfølgning. Mens den blev observeret på tværs af alle aldersgrupper, så den højeste risiko for nedsat højde ved slutningen af langtidsopfølgningen ud til at korrelere med påbegyndelse af kombinationsbehandling i årene med forventet maksimal væksthastighed. [Se ADVARSLER OG FORHOLDSREGLER ]
Laboratorieværdier
Voksne og pædiatriske emner
Bivirkningsprofilen i undersøgelse 3, som sammenlignede PegIntron/vægt-baseret REBETOL 200 mg kombination med en PegIntron/flad dosis REBETOL 200 mg regime, afslørede en øget forekomst af anæmi med vægtbaseret dosering (29 % vs. 19 % for vægtbaseret) vs. flad dosis regimer, henholdsvis). Imidlertid var størstedelen af tilfældene af anæmi milde og reagerede på dosisreduktioner.
Ændringer i udvalgte laboratorieværdier under behandling i kombination med REBETOL 200 mg behandling er beskrevet nedenfor. Fald i hæmoglobin, leukocytter, neutrofiler og blodplader kan kræve dosisreduktion eller permanent seponering af behandlingen [se DOSERING OG ADMINISTRATION ]. Ændringer i udvalgte laboratorieværdier under behandlingen er beskrevet i tabel 8. De fleste af ændringerne i laboratorieværdier i PegIntron/REBETOL 200 mg-studiet med pædiatri var milde eller moderate.
Hæmoglobin
Hæmoglobinniveauerne faldt til mindre end 11 g/dL hos omkring 30 % af forsøgspersonerne i undersøgelse 2. I undersøgelse 3 havde 47 % af forsøgspersonerne, der fik WBD REBETOL 200 mg og 33 % på fladdosis REBETOL 200 mg, fald i hæmoglobinniveauet på mindre end 11 g. /dl. Reduktioner i hæmoglobin til mindre end 9 g/dL forekom hyppigere hos forsøgspersoner, der fik WBD sammenlignet med flad dosering (henholdsvis 4 % og 2 %). I undersøgelse 2 var dosisændring påkrævet hos 9 % og 13 % af forsøgspersonerne i grupperne PegIntron/REBETOL 200 mg og INTRON A/REBETOL 200 mg. I undersøgelse 4 havde forsøgspersoner, der fik PegIntron (1,5 mcg/kg)/REBETOL fald i hæmoglobinniveauet til mellem 8,5 og mindre end 10 g/dL (28 %) og til mindre end 8,5 g/dL (3 %), hvorimod hos patienter Da de fik Pegasys 180 mcg/Copegus, forekom disse fald hos henholdsvis 26 % og 4 % af forsøgspersonerne. Hæmoglobinniveauet blev stabilt ved behandlingsuge 4-6 i gennemsnit. Det typiske mønster, der blev observeret, var et fald i hæmoglobinniveauet ved behandlingsuge 4 efterfulgt af stabilisering og et plateau, som blev opretholdt til slutningen af behandlingen. I PegIntron-monoterapiforsøget var hæmoglobinfald generelt milde, og dosisændringer var sjældent nødvendige [se DOSERING OG ADMINISTRATION ].
Neutrofiler
Fald i neutrofiltal blev observeret hos et flertal af voksne forsøgspersoner behandlet med kombinationsbehandling med REBETOL 200 mg i undersøgelse 2 (85 %) og INTRON A/REBETOL (60 %). Alvorlig potentielt livstruende neutropeni (mindre end 0,5 x 109/L) forekom hos 2 % af forsøgspersonerne behandlet med INTRON A/REBETOL 200 mg og hos ca. 4 % af forsøgspersonerne behandlet med PegIntron/REBETOL 200 mg i undersøgelse 2. 18 procent af forsøgspersonerne. PegIntron/REBETOL i undersøgelse 2 krævede ændring af interferon-dosis. Få forsøgspersoner (mindre end 1%) krævede permanent seponering af behandlingen. Neutrofiltallet vendte generelt tilbage til niveauet før behandling 4 uger efter ophør af behandlingen [se DOSERING OG ADMINISTRATION ].
Blodplader
Trombocyttallet faldt til mindre end 100.000/mm³ hos ca. 20 % af forsøgspersonerne behandlet med PegIntron alene eller med REBETOL 200 mg og hos 6 % af voksne forsøgspersoner behandlet med INTRON A/REBETOL. Alvorlige fald i blodpladetal (mindre end 50.000/mm³) forekommer hos mindre end 4 % af voksne forsøgspersoner. Patienter kan have behov for seponering eller dosisændring som følge af blodpladefald [se DOSERING OG ADMINISTRATION ]. I undersøgelse 2 krævede 1 % eller 3 % af forsøgspersonerne dosisændring af henholdsvis INTRON A eller PegIntron. Trombocyttallet vendte generelt tilbage til niveauerne før behandling 4 uger efter behandlingens ophør.
Skjoldbruskkirtel funktion
Udvikling af TSH-abnormiteter, med eller uden kliniske manifestationer, er forbundet med interferonbehandlinger. I undersøgelse 2 forekom klinisk tilsyneladende skjoldbruskkirtellidelser blandt forsøgspersoner behandlet med enten INTRON A eller PegIntron (med eller uden REBETOL) med en lignende forekomst (5 % for hypothyroidisme og 3 % for hyperthyroidisme). Forsøgspersonerne udviklede nye TSH-abnormiteter under behandling og i opfølgningsperioden. Ved slutningen af opfølgningsperioden havde 7 % af forsøgspersonerne stadig unormale TSH-værdier.
Bilirubin og urinsyre
I undersøgelse 2 udviklede 10 til 14 % af forsøgspersonerne hyperbilirubinæmi, og 33 til 38 % udviklede hyperurikæmi i forbindelse med hæmolyse. Seks forsøgspersoner udviklede mild til moderat gigt.
Erfaring med kliniske forsøg – REBETOL/INTRON En kombinationsterapi
Voksne emner
kliniske forsøg ophørte henholdsvis 19 % og 6 % af tidligere ubehandlede og tilbagefaldspersoner behandlingen på grund af bivirkninger i kombinationsarmene sammenlignet med 13 % og 3 % i interferonarmene. Udvalgte behandlingsrelaterede bivirkninger, der opstod i de amerikanske forsøg med en forekomst på mere end eller lig med 5 %, er angivet efter behandlingsgruppe (se tabel 9). Generelt blev de udvalgte behandlingsrelaterede bivirkninger rapporteret med lavere forekomst i de internationale forsøg sammenlignet med de amerikanske forsøg, med undtagelse af asteni, influenzalignende symptomer, nervøsitet og kløe.
Pædiatriske emner
kliniske forsøg med 118 pædiatriske forsøgspersoner i alderen 3 til 16 år afbrød 6 % behandlingen på grund af bivirkninger. Dosisændringer var påkrævet hos 30 % af forsøgspersonerne, oftest for anæmi og neutropeni. Generelt svarede bivirkningsprofilen i den pædiatriske population til den, der blev observeret hos voksne. Forstyrrelser på injektionsstedet, feber, anoreksi, opkastning og følelsesmæssig labilitet forekom hyppigere hos pædiatriske forsøgspersoner sammenlignet med voksne. Omvendt oplevede pædiatriske forsøgspersoner mindre træthed, dyspepsi, artralgi, søvnløshed, irritabilitet, nedsat koncentrationsevne, dyspnø og kløe sammenlignet med voksne forsøgspersoner. Udvalgte behandlingsrelaterede bivirkninger, der forekom med større end eller lig med 5 % forekomst blandt alle pædiatriske forsøgspersoner, som fik den anbefalede dosis af REBETOL/INTRON A kombinationsbehandling, er angivet i tabel 9.
løbet af et 48-ugers behandlingsforløb var der et fald i hastigheden af lineær vækst (gennemsnitlig percentiltildelingsfald på 7%) og et fald i hastigheden af vægtøgning (gennemsnitlig percentiltildelingsfald på 9%). En generel vending af disse tendenser blev bemærket i løbet af den 24-ugers efterbehandlingsperiode. Langtidsdata fra et begrænset antal patienter tyder dog på, at kombinationsbehandling kan inducere en væksthæmning, der resulterer i reduceret endelig voksenhøjde hos nogle patienter [se ADVARSLER OG FORHOLDSREGLER ].
Laboratorieværdier
Ændringer i udvalgte hæmatologiske værdier (hæmoglobin, hvide blodlegemer, neutrofiler og blodplader) under behandlingen er beskrevet nedenfor (se tabel 10).
Hæmoglobin. Hæmoglobinfald blandt forsøgspersoner, der modtog REBETOL-behandling, begyndte ved uge 1, med stabilisering i uge 4. Hos tidligere ubehandlede forsøgspersoner behandlet i 48 uger var det gennemsnitlige maksimale fald fra baseline 3,1 g/dL i det amerikanske forsøg og 2,9 g/dL i det internationale forsøg. forsøg. Hos patienter med tilbagefald var det gennemsnitlige maksimale fald fra baseline 2,8 g/dL i det amerikanske forsøg og 2,6 g/dL i det internationale forsøg. Hæmoglobinværdierne vendte tilbage til niveauerne før behandling inden for 4 til 8 uger efter behandlingens ophør hos de fleste forsøgspersoner.
Bilirubin og urinsyre. Forøgelser i både bilirubin og urinsyre, forbundet med hæmolyse, blev noteret i kliniske forsøg. De fleste var moderate biokemiske ændringer og blev vendt inden for 4 uger efter seponering af behandlingen. Denne observation forekom hyppigst hos personer med en tidligere diagnose af Gilberts syndrom. Dette har ikke været forbundet med leverdysfunktion eller klinisk morbiditet.
Postmarketing-oplevelser
Følgende bivirkninger er blevet identificeret og rapporteret under brug af REBETOL efter godkendelse i kombination med INTRON A eller PegIntron. Fordi disse reaktioner rapporteres frivilligt fra en population af usikker størrelse, er det ikke altid muligt pålideligt at estimere deres hyppighed eller etablere en årsagssammenhæng til lægemiddeleksponering.
Blod og lymfesystem lidelser
Pure red cell aplasia, aplastisk anæmi
Øre- og labyrintlidelser
Høreforstyrrelser, svimmelhed
Åndedræts-, thorax- og mediastinale lidelser
Pulmonal hypertension
Øjenlidelser
Serøs nethindeløsning
Endokrine lidelser
Diabetes
DRUGSINTERAKTIONER
Didanosin
Eksponering for didanosin eller dets aktive metabolit (dideoxyadenosin 5'-triphosphat) øges, når didanosin administreres sammen med ribavirin, hvilket kan forårsage eller forværre klinisk toksicitet; Derfor er samtidig administration af REBETOL 200 mg kapsler eller oral opløsning og didanosin kontraindiceret. Rapporter om fatalt leversvigt, såvel som perifer neuropati, pancreatitis og symptomatisk hyperlaktæmi/laktatacidose er blevet rapporteret i kliniske forsøg.
Nukleosid-analoger
Leverdekompensation (nogle dødelig) er forekommet hos cirrose HIV/HCV co-inficerede patienter, der modtager antiretroviral kombinationsbehandling for HIV og interferon alfa og ribavirin. Tilføjelse af behandling med alfa-interferoner alene eller i kombination med ribavirin kan øge risikoen i denne patientpopulation. Patienter, der får interferon med ribavirin og nukleosid revers transkriptasehæmmere (NRTI'er), bør overvåges nøje for behandlingsassocierede toksiciteter, især leverdekompensation og anæmi. Seponering af NRTI'er bør betragtes som medicinsk passende (se mærkning for individuelle NRTI-produkter ). Dosisreduktion eller seponering af interferon, ribavirin eller begge bør også overvejes, hvis der observeres forværrede kliniske toksiciteter, herunder leverdekompensation (f.eks. Child-Pugh større end 6).
Ribavirin kan antagonisere cellekulturens antivirale aktivitet af stavudin og zidovudin mod HIV. Ribavirin har vist sig i cellekultur at hæmme phosphorylering af lamivudin, stavudin og zidovudin, hvilket kan føre til nedsat antiretroviral aktivitet. I et studie med et andet pegyleret interferon i kombination med ribavirin blev der imidlertid ikke observeret nogen farmakokinetisk (f.eks. plasmakoncentrationer eller intracellulære triphosphorylerede aktive metabolitkoncentrationer) eller farmakodynamisk (f.eks. tab af HIV/HCV virologisk undertrykkelse) interaktion, når ribavirin og lamivudin (n) =18), stavudin (n=10) eller zidovudin (n=6) blev administreret samtidigt som en del af et multilægemiddelregime til HIV/HCV co-inficerede forsøgspersoner. Derfor bør samtidig brug af ribavirin med et af disse lægemidler anvendes med forsigtighed.
Lægemidler metaboliseret af Cytochrom P-450
Resultater af in vitro-undersøgelser med både humane og rottelevermikrosompræparater indikerede ringe eller ingen cytokrom P-450 enzymmedieret metabolisme af ribavirin med minimalt potentiale for P-450 enzymbaserede lægemiddelinteraktioner.
Der blev ikke observeret nogen farmakokinetiske interaktioner mellem INTRON A og REBETOL 200 mg kapsler i et farmakokinetisk studie med flere doser.
Azathioprin
Brug af ribavirin til behandling af kronisk hepatitis C hos patienter, der får azathioprin, er blevet rapporteret at inducere alvorlig pancytopeni og kan øge risikoen for azathioprin-relateret myelotoksicitet. Inosinmonophosphatdehydrogenase (IMDH) er nødvendig for en af azathioprins metaboliske veje. Ribavirin er kendt for at hæmme IMDH, hvilket fører til akkumulering af en azathioprin-metabolit, 6-methylthioinosinmonophosphat (6-MTITP), som er forbundet med myelotoksicitet (neutropeni, trombocytopeni og anæmi). Patienter, der får azathioprin sammen med ribavirin, bør have fuldstændige blodtællinger, inklusive trombocyttal, overvåget ugentligt i den første måned, to gange om måneden i den anden og tredje måned af behandlingen, derefter månedligt eller oftere, hvis dosis eller andre terapiændringer er nødvendige [se ADVARSLER OG FORHOLDSREGLER ].
ADVARSLER
Inkluderet som en del af FORHOLDSREGLER afsnit.
FORHOLDSREGLER
Graviditet
REBETOL 200mg kapsler og oral opløsning kan forårsage fosterskader og død hos det ufødte barn. Behandling med REBETOL bør ikke påbegyndes, før der er modtaget en rapport om en negativ graviditetstest umiddelbart før planlagt påbegyndelse af behandlingen. Patienter bør bruge mindst to former for prævention og have månedlige graviditetstest under behandlingen og i 6-månedersperioden efter behandlingen er stoppet. Der skal udvises ekstrem forsigtighed for at undgå graviditet hos kvindelige patienter og hos mandlige patienters kvindelige partnere. REBETOL 200mg har vist signifikante teratogene og embryocidale virkninger hos alle dyrearter, hvor der er udført tilstrækkelige undersøgelser. Disse virkninger opstod ved doser som lavt som en tyvendedel af den anbefalede humane dosis af ribavirin. Behandling med REBETOL 200 mg bør ikke påbegyndes før en rapport om en negativ graviditetstest er indhentet umiddelbart før planlagt behandlingsstart [se KASSET ADVARSEL , KONTRAINDIKATIONER , Brug i specifikke populationer , og PATIENTOPLYSNINGER ].
Anæmi
Den primære toksicitet af ribavirin er hæmolytisk anæmi, som blev observeret hos ca. 10 % af REBETOL/INTRON A-behandlede forsøgspersoner i kliniske forsøg. Anæmien forbundet med REBETOL kapsler opstår inden for 1 til 2 uger efter påbegyndelse af behandlingen. Da det indledende fald i hæmoglobin kan være signifikant, tilrådes det, at hæmoglobin eller hæmatokrit opnås før behandlingsstart og i uge 2 og uge 4 af behandlingen, eller oftere, hvis det er klinisk indiceret. Patienterne skal derefter følges efter klinisk passende [se DOSERING OG ADMINISTRATION ].
Fatale og ikke-fatale myokardieinfarkter er blevet rapporteret hos patienter med anæmi forårsaget af REBETOL. Patienter bør vurderes for underliggende hjertesygdom før påbegyndelse af ribavirinbehandling. Patienter med allerede eksisterende hjertesygdom bør have elektrokardiogrammer indgivet før behandling og bør overvåges passende under behandlingen. Hvis der er nogen forværring af kardiovaskulær status, bør behandlingen suspenderes eller seponeres [se DOSERING OG ADMINISTRATION ]. Fordi hjertesygdom kan forværres af lægemiddelinduceret anæmi, bør patienter med en historie med signifikant eller ustabil hjertesygdom ikke bruge REBETOL.
Pancreatitis
Behandling med REBETOL og INTRON A eller PegIntron bør seponeres hos patienter med tegn og symptomer på pancreatitis og seponeres hos patienter med bekræftet pancreatitis.
Lungesygdomme
Lungesymptomer, herunder dyspnø, pulmonære infiltrater, pneumonitis, pulmonal hypertension og lungebetændelse, er blevet rapporteret under behandling med REBETOL med alfa-interferon kombinationsbehandling; lejlighedsvise tilfælde af dødelig lungebetændelse er forekommet. Derudover er sarkoidose eller forværring af sarkoidose blevet rapporteret. Hvis der er tegn på lungeinfiltrater eller nedsat lungefunktion, skal patienten overvåges nøje, og hvis det er relevant, skal kombinationsbehandlingen seponeres.
Oftalmologiske lidelser
Ribavirin bruges i kombinationsbehandling med alfa-interferoner. Nedsat eller tab af syn, retinopati inklusive makulært ødem, retinal arterie eller vene, trombose, nethindeblødninger og vatpletter, optisk neuritis, papilleødem og serøs nethindeløsning induceres eller forværres ved behandling med alfa-interferoner. Alle patienter bør få en øjenundersøgelse ved baseline. Patienter med allerede eksisterende oftalmologiske lidelser (f.eks. diabetisk eller hypertensiv retinopati) bør få periodiske oftalmologiske undersøgelser under kombinationsbehandling med alfa-interferonbehandling. Enhver patient, der udvikler øjensymptomer, bør få en hurtig og fuldstændig øjenundersøgelse. Kombinationsbehandling med alfa-interferoner bør seponeres hos patienter, som udvikler nye eller forværrede oftalmologiske lidelser.
Laboratorieprøver
PegIntron i kombination med ribavirin kan forårsage alvorlige fald i neutrofil- og blodpladetal samt hæmatologiske, endokrine (f.eks. TSH) og leverabnormiteter.
Patienter i kombinationsbehandling med PegIntron/REBETOL skal have hæmatologi- og blodkemitestning før påbegyndelse af behandlingen og derefter periodisk derefter. I det kliniske forsøg til voksne blev fuldstændige blodtal (inklusive hæmoglobin-, neutrofil- og blodpladetal) og kemi (inklusive AST, ALT, bilirubin og urinsyre) målt under behandlingsperioden i uge 2, 4, 8, 12 og derefter med 6 ugers mellemrum, eller hyppigere, hvis der udvikles abnormiteter. Hos pædiatriske forsøgspersoner blev de samme laboratorieparametre evalueret med yderligere vurdering af hæmoglobin ved behandlingsuge 6. TSH-niveauer blev målt hver 12. uge i behandlingsperioden. HCV-RNA bør måles periodisk under behandlingen [se DOSERING OG ADMINISTRATION ].
Tand- og parodontale lidelser
Dentale og parodontale lidelser er blevet rapporteret hos patienter, der får kombinationsbehandling med ribavirin og interferon eller peginterferon. Derudover kan mundtørhed have en skadelig effekt på tænder og slimhinder i munden under langtidsbehandling med kombinationen af REBETOL og pegyleret eller ikke-pegyleret interferon alfa-2b. Patienter bør børste deres tænder grundigt to gange dagligt og have regelmæssige tandundersøgelser. Hvis der opstår opkastning, skal de rådes til at skylle munden grundigt ud bagefter.
Samtidig administration af Azathioprin
Pancytopeni (markant fald i røde blodlegemer, neutrofiler og blodplader) og knoglemarvssuppression er blevet rapporteret i litteraturen for at forekomme inden for 3 til 7 uger efter samtidig administration af pegyleret interferon/ribavirin og azathioprin. Hos dette begrænsede antal patienter (n=8) var myelotoksiciteten reversibel inden for 4 til 6 uger efter seponering af både HCV antiviral behandling og samtidig azathioprin og opstod ikke igen ved genindførelse af nogen af behandlingen alene. PegIntron, REBETOL og azathioprin bør seponeres for pancytopeni, og pegyleret interferon/ribavirin bør ikke genintroduceres med samtidig azathioprin [se DRUGSINTERAKTIONER ].
Indvirkning på vækst - pædiatrisk brug
Data om virkningerne af PegIntron og REBETOL på væksten kommer fra et åbent studie med forsøgspersoner i alderen 3 til 17 år, hvor vægt- og højdeændringer sammenlignes med amerikanske normative befolkningsdata. Generelt halter vægt- og højdeforøgelsen hos pædiatriske forsøgspersoner behandlet med PegIntron og REBETOL 200 mg bagud, hvad der er forudsagt af normative populationsdata for hele behandlingens længde. Svært hæmmet væksthastighed (mindre end 3. percentil) blev observeret hos 70 % af forsøgspersonerne under behandling. Efter behandling forekom rebound-vækst og vægtøgning hos de fleste forsøgspersoner. Langtidsopfølgningsdata hos pædiatriske forsøgspersoner indikerer dog, at PegIntron i kombinationsbehandling med REBETOL kan inducere en væksthæmning, der resulterer i nedsat voksenhøjde hos nogle patienter [se BIVIRKNINGER ].
Tilsvarende blev der set en påvirkning af væksten hos forsøgspersoner efter behandling med REBETOL 200 mg og INTRON A kombinationsbehandling i et år. I et langsigtet opfølgningsforsøg med et begrænset antal af disse forsøgspersoner resulterede kombinationsbehandling i reduceret endelig voksenhøjde hos nogle forsøgspersoner [se BIVIRKNINGER ].
Sikkerhedsforanstaltninger ved brug
Baseret på resultater af kliniske forsøg er ribavirin monoterapi ikke effektiv til behandling af kronisk hepatitis C-virusinfektion; derfor må REBETOL 200mg kapsler eller oral opløsning ikke anvendes alene. Sikkerheden og effekten af REBETOL kapsler og oral opløsning er kun blevet fastslået, når de anvendes sammen med INTRON A eller PegIntron (ikke andre interferoner) som kombinationsbehandling.
Sikkerheden og effekten af behandling med REBETOL/INTRON A og PegIntron til behandling af HIV-infektion, adenovirus, RSV, parainfluenza eller influenzainfektioner er ikke blevet fastlagt. REBETOL 200mg kapsler bør ikke anvendes til disse indikationer. Ribavirin til inhalation har separat mærkning, som bør konsulteres, hvis ribavirin inhalationsbehandling overvejes.
Der er betydelige bivirkninger forårsaget af behandling med REBETOL/INTRON A eller PegIntron, herunder svær depression og selvmordstanker, hæmolytisk anæmi, undertrykkelse af knoglemarvsfunktion, autoimmune og infektionssygdomme, pulmonal dysfunktion, pancreatitis og diabetes. Selvmordstanker eller selvmordsforsøg forekom hyppigere blandt pædiatriske patienter, primært unge, sammenlignet med voksne patienter (2,4 % versus 1 %) under behandling og opfølgning uden for behandlingen. Mærkning af INTRON A og PegIntron bør gennemgås i deres helhed for at få yderligere sikkerhedsoplysninger før påbegyndelse af kombinationsbehandling.
Patientrådgivningsinformation
Råd patienten til at læse den FDA-godkendte patientmærkning ( Medicinvejledning ).
Anæmi
Den mest almindelige uønskede oplevelse med REBETOL kapsler er anæmi, som kan være alvorlig [se ADVARSLER OG FORHOLDSREGLER og BIVIRKNINGER ]. Patienter bør informeres om, at laboratorieevalueringer er påkrævet før påbegyndelse af behandlingen og periodisk derefter [se DOSERING OG ADMINISTRATION ]. Det tilrådes, at patienterne er godt hydrerede, især i de indledende stadier af behandlingen.
Graviditet
Patienterne skal informeres om, at REBETOL 200 mg kapsler og oral opløsning kan forårsage fosterskader og døden hos det ufødte barn. REBETOL 200mg må ikke anvendes af kvinder, der er gravide eller af mænd, hvis kvindelige partnere er gravide. Der skal udvises ekstrem forsigtighed for at undgå graviditet hos kvindelige patienter og hos kvindelige partnere til mandlige patienter, der tager REBETOL. REBETOL bør ikke påbegyndes, før der er indhentet en rapport om en negativ graviditetstest umiddelbart før påbegyndelse af behandlingen. Patienter skal udføre en graviditetstest månedligt under behandlingen og i 6 måneder efter behandlingen.
Kvinder i den fødedygtige alder skal rådgives om brug af effektiv prævention (to pålidelige former) før påbegyndelse af behandlingen. Patienter (mænd og kvinder) skal informeres om de teratogene/embryocidale risici og skal instrueres i at anvende effektiv prævention under REBETOL 200 mg og i 6 måneder efter behandling. Patienter (mænd og kvinder) bør rådes til straks at underrette lægen i tilfælde af graviditet [se KONTRAINDIKATIONER , ADVARSLER OG FORHOLDSREGLER , og Brug i specifikke populationer ].
Hvis graviditet opstår under behandling eller i løbet af 6 måneder efter behandling, skal patienten informeres om den teratogene risiko ved behandling med REBETOL 200 mg for fosteret. Patienter eller partnere til patienter skal straks rapportere enhver graviditet, der opstår under behandlingen eller inden for 6 måneder efter behandlingens ophør, til deres læge. Udskrivende læger bør rapportere sådanne tilfælde ved at ringe til 1-800-593-2214.
Risici versus fordele
Patienter, der får REBETOL-kapsler, skal informeres om fordele og risici forbundet med behandlingen, instrueres i dens passende brug og henvises til patienten. Medicinvejledning . Patienterne skal informeres om, at virkningen af behandling af hepatitis C-infektion på overførsel ikke er kendt, og at passende forholdsregler for at forhindre overførsel af hepatitis C-virus bør træffes.
Patienterne skal informeres om, hvad de skal gøre, hvis de glemmer en dosis af REBETOL; den glemte dosis skal tages så hurtigt som muligt i løbet af samme dag. Patienter bør ikke fordoble den næste dosis. Patienter bør rådes til at kontakte deres læge, hvis de har spørgsmål.
Ikke-klinisk toksikologi
Karcinogenese, mutagenese, svækkelse af fertilitet
Karcinogenese
Ribavirin forårsagede ikke en stigning i nogen tumortype, når det blev administreret i 6 måneder i den transgene p53-deficiente musemodel i doser op til 300 mg/kg (estimeret human ækvivalent på 25 mg/kg baseret på kropsoverfladejustering for en voksen på 60 kg 1,9 gange den maksimale anbefalede daglige dosis til mennesker). Ribavirin var ikke-karcinogent, når det blev administreret i 2 år til rotter i doser op til 40 mg/kg (estimeret human ækvivalent på 5,71 mg/kg baseret på kropsoverfladejustering for en voksen på 60 kg).
Mutagenese
Ribavirin viste øget forekomst af mutation og celletransformation i flere genotoksicitetsassays. Ribavirin var aktiv i Balb/3T3 in vitro celletransformationsanalysen. Mutagen aktivitet blev observeret i muselymfomanalysen og ved doser på 20 til 200 mg/kg (estimeret human ækvivalent på 1,67 til 16,7 mg/kg, baseret på kropsoverfladejustering for en voksen på 60 kg; 0,1 til 1 gange maksimum anbefalet human 24-timers dosis af ribavirin) i en mikronukleusanalyse hos mus. Et dominant letalt assay hos rotter var negativt, hvilket indikerer, at hvis der opstod mutationer i rotter, blev de ikke overført gennem hankønsceller.
Forringelse af fertilitet
Ribavirin viste signifikante embryocidale og teratogene virkninger ved doser langt under den anbefalede humane dosis hos alle dyrearter, hvor der er udført tilstrækkelige undersøgelser. Misdannelser af kraniet, ganen, øjet, kæben, lemmerne, skelettet og mave-tarmkanalen blev noteret. Hyppigheden og sværhedsgraden af teratogene virkninger steg med eskalering af lægemiddeldosis. Overlevelsen af fostre og afkom blev reduceret. I konventionelle embryotoksicitets-/teratogenicitetsundersøgelser hos rotter og kaniner var dosisniveauer uden effekt et godt stykke under niveauet for foreslået klinisk anvendelse (0,3 mg/kg/dag for både rotte og kanin; ca. 0,06 gange den anbefalede humane 24-timers dosis af ribavirin). Ingen maternel toksicitet eller virkninger på afkom blev observeret i et peri/postnatalt toksicitetsstudie i rotter doseret oralt med op til 1 mg/kg/dag (estimeret human ækvivalent dosis på 0,17 mg/kg baseret på kropsoverfladejustering for en 60 kg voksen voksen 0,01 gange den maksimalt anbefalede humane 24-timers dosis af ribavirin) [se KONTRAINDIKATIONER , og ADVARSLER OG FORHOLDSREGLER ].
Fertile kvinder og partnere til fertile kvinder bør ikke få REBETOL, medmindre patienten og hans/hendes partner bruger effektiv prævention (to pålidelige former). Baseret på en flerdosis-halveringstid (t1/2) af ribavirin på 12 dage, skal der anvendes effektiv prævention i 6 måneder efter behandlingen (f.eks. 15 halveringstider for clearance for ribavirin).
REBETOL bør anvendes med forsigtighed til fertile mænd. I undersøgelser på mus for at evaluere tidsforløbet og reversibiliteten af ribavirin-induceret testikeldegeneration ved doser på 15 til 150 mg/kg/dag (estimeret human ækvivalent på 1,25 til 12,5 mg/kg/dag, baseret på kropsoverfladejustering for en 60 kg voksen; 0,1-0,8 gange den maksimale humane 24-timers dosis af ribavirin) administreret i 3 eller 6 måneder, forekom abnormiteter i sædceller. Ved ophør af behandlingen var i det væsentlige total genopretning fra ribavirin-induceret testikeltoksicitet synlig inden for 1 eller 2 spermatogenesecyklusser.
Brug i specifikke populationer
Graviditet
Graviditetskategori X
[Se KONTRAINDIKATIONER , ADVARSLER OG FORHOLDSREGLER , og Ikke-klinisk toksikologi ].
Behandling og efterbehandling:
Potentiel risiko for fosteret
Ribavirin er kendt for at akkumulere i intracellulære komponenter, hvorfra det udskilles meget langsomt. Det vides ikke, om ribavirin indeholdt i sædceller vil udøve en potentiel teratogene virkning ved befrugtning af æggene. I en undersøgelse med rotter blev det konkluderet, at dominant dødelighed ikke blev induceret af ribavirin ved doser på op til 200 mg/kg i 5 dage (estimerede humane ækvivalente doser på 7,14 til 28,6 mg/kg, baseret på kropsoverfladejustering for 60 kg voksen; op til 1,7 gange den maksimalt anbefalede humane dosis af ribavirin). Men på grund af de potentielle humane teratogene virkninger af ribavirin, bør mandlige patienter rådes til at tage alle forholdsregler for at undgå risiko for graviditet for deres kvindelige partnere.
Kvinder i den fødedygtige alder bør ikke få REBETOL, medmindre de bruger effektiv prævention (to pålidelige former) i terapiperioden. Derudover bør effektiv prævention anvendes i 6 måneder efter behandling baseret på en halveringstid for flere doser (t1/2) af ribavirin på 12 dage.
Mandlige patienter og deres kvindelige partnere skal praktisere effektiv prævention (to pålidelige former) under behandling med REBETOL og i 6-måneders perioden efter behandling (f.eks. 15 halveringstider for ribavirin-clearance fra kroppen).
Et Ribavirin-graviditetsregister er blevet etableret for at overvåge moderens-føtale resultater af graviditeter hos kvindelige patienter og kvindelige partnere til mandlige patienter, der er blevet eksponeret for ribavirin under behandlingen og i 6 måneder efter behandlingens ophør. Læger og patienter opfordres til at rapportere sådanne tilfælde ved at ringe på 1-800-593-2214.
Ammende mødre
Det vides ikke, om REBETOL-produktet udskilles i modermælk. På grund af risikoen for alvorlige bivirkninger fra lægemidlet hos ammende spædbørn, bør der træffes en beslutning om, hvorvidt amningen skal afbrydes eller om at udskyde eller seponere REBETOL.
Pædiatrisk brug
Sikkerhed og effektivitet af REBETOL 200 mg i kombination med PegIntron er ikke blevet fastslået hos pædiatriske patienter under 3 år. Ved behandling med REBETOL/INTRON A bør tegn på sygdomsprogression, såsom leverbetændelse og fibrose, samt prognostiske faktorer for respons, HCV-genotype og viral belastning tages i betragtning, når det besluttes at behandle en pædiatrisk patient. Fordelene ved behandlingen skal afvejes mod de observerede sikkerhedsresultater.
Langtidsopfølgningsdata hos pædiatriske forsøgspersoner indikerer, at REBETOL i kombination med PegIntron eller med INTRON A kan inducere en væksthæmning, der resulterer i nedsat højde hos nogle patienter [se ADVARSLER OG FORHOLDSREGLER og BIVIRKNINGER ].
Selvmordstanker eller selvmordsforsøg forekom hyppigere blandt pædiatriske patienter, primært teenagere, sammenlignet med voksne patienter (2,4 % vs. 1 %) under behandling og opfølgning uden for behandlingen [se ADVARSLER OG FORHOLDSREGLER ]. Som hos voksne patienter oplevede pædiatriske patienter andre psykiatriske bivirkninger (f.eks. depression, følelsesmæssig labilitet, somnolens), anæmi og neutropeni [se ADVARSLER OG FORHOLDSREGLER ].
Geriatrisk brug
Kliniske forsøg med REBETOL/INTRON A- eller PegIntron-behandling omfattede ikke et tilstrækkeligt antal forsøgspersoner på 65 år og derover til at afgøre, om de reagerer anderledes end yngre forsøgspersoner.
REBETOL 200mg er kendt for at udskilles væsentligt af nyrerne, og risikoen for toksiske reaktioner på dette lægemiddel kan være større hos patienter med nedsat nyrefunktion. Da ældre patienter ofte har nedsat nyrefunktion, bør der udvises forsigtighed ved dosisvalg. Nyrefunktionen bør overvåges, og dosisjusteringer bør foretages i overensstemmelse hermed. REBETOL 200mg bør ikke anvendes til patienter med kreatininclearance mindre end 50 ml/min [se KONTRAINDIKATIONER ].
Generelt bør REBETOL 200 mg kapsler administreres til ældre patienter med forsigtighed, begyndende i den nedre ende af doseringsområdet, hvilket afspejler den større hyppighed af nedsat lever- og hjertefunktion og af samtidig sygdom eller anden lægemiddelbehandling. I kliniske forsøg havde ældre forsøgspersoner en højere frekvens af anæmi (67 %) end yngre patienter (28 %) [se ADVARSLER OG FORHOLDSREGLER ].
Organtransplantationsmodtagere
Sikkerheden og effekten af INTRON A og PegIntron alene eller i kombination med REBETOL til behandling af hepatitis C hos lever- eller andre organtransplanterede modtagere er ikke blevet fastlagt. I et lille (n=16) enkeltcenter, ukontrolleret tilfælde, var nyresvigt hos nyreallotransplanterede modtagere, der fik interferon alfa og ribavirin kombinationsbehandling, hyppigere end forventet ud fra centrets tidligere erfaring med nyreallotransplanterede modtagere, der ikke fik kombinationsbehandling. Forholdet mellem nyresvigt og renal allotransplantatafstødning er ikke klart.
HIV eller HBV Co-infektion
Sikkerheden og effekten af PegIntron/REBETOL 200 mg og INTRON A/REBETOL til behandling af patienter med HCV co-inficeret med HIV eller HBV er ikke blevet fastlagt.
OVERDOSIS
Der er begrænset erfaring med overdosering. Akut indtagelse af op til 20 g REBETOL-kapsler, INTRON A-indtagelse på op til 120 millioner enheder og subkutane doser af INTRON A op til 10 gange de anbefalede doser er blevet rapporteret. Primære virkninger, der er blevet observeret, er øget forekomst og sværhedsgrad af bivirkninger relateret til den terapeutiske brug af INTRON A og REBETOL. Leverenzymabnormaliteter, nyresvigt, blødning og myokardieinfarkt er imidlertid blevet rapporteret ved administration af enkelt subkutane doser af INTRON A, der overstiger doseringsanbefalingerne.
Der er ingen specifik modgift mod INTRON A eller REBETOL 200 mg overdosis, og hæmodialyse og peritonealdialyse er ikke effektive til behandling af overdosering af disse midler.
KONTRAINDIKATIONER
REBETOL kombinationsbehandling er kontraindiceret ved:
- kvinder, der er gravide. REBETOL 200mg kan forårsage fosterskader, når det administreres til en gravid kvinde. REBETOL er kontraindiceret til kvinder, der er eller kan blive gravide. Hvis REBETOL bruges under graviditet, eller hvis patienten bliver gravid, mens hun tager REBETOL 200 mg, skal patienten informeres om den potentielle fare for hendes foster [se ADVARSLER OG FORHOLDSREGLER , Brug i specifikke populationer , og PATIENTOPLYSNINGER ]
- mænd, hvis kvindelige partnere er gravide
- patienter med kendte overfølsomhedsreaktioner såsom Stevens-Johnsons syndrom, toksisk, epidermal nekrolyse og erythema multiforme over for ribavirin eller en hvilken som helst komponent i produktet
- patienter med autoimmun hepatitis
- patienter med hæmoglobinopatier (f.eks. thalassæmi major, seglcelleanæmi)
- patienter med kreatininclearance mindre end 50 ml/min. [se Brug i specifikke populationer og KLINISK FARMAKOLOGI ]
- Samtidig administration af REBETOL og didanosin er kontraindiceret, fordi eksponeringen for den aktive metabolit af didanosin (dideoxyadenosin 5'-triphosphat) er øget. Fatalt leversvigt såvel som perifer neuropati, pancreatitis og symptomatisk hyperlactatæmi/laktatacidose er blevet rapporteret hos patienter, der får didanosin i kombination med ribavirin [se DRUGSINTERAKTIONER ].
KLINISK FARMAKOLOGI
Handlingsmekanisme
Ribavirin er et antiviralt middel [se Mikrobiologi ].
Farmakokinetik
Enkelt- og multiple-dosis farmakokinetiske egenskaber hos voksne er opsummeret i tabel 11. Ribavirin blev hurtigt og omfattende absorberet efter oral administration. Men på grund af first-pass metabolisme var den absolutte biotilgængelighed i gennemsnit 64 % (44 %). Der var en lineær sammenhæng mellem dosis og AUCtf (AUC fra tidspunkt nul til sidste målbare koncentration) efter enkeltdoser på 200 til 1200 mg ribavirin. Forholdet mellem dosis og Cmax var krumlinjet og havde tendens til at asymptotere over enkeltdoser på 400 til 600 mg.
Ved flere orale doseringer, baseret på AUC12 timer, blev der observeret en 6-fold akkumulering af ribavirin i plasma. Efter oral dosering med 600 mg to gange dagligt blev steady-state nået efter ca. 4 uger med gennemsnitlige steady-state plasmakoncentrationer på 2200 ng/ml (37%). Efter seponering af doseringen var den gennemsnitlige halveringstid 298 (30 %) timer, hvilket sandsynligvis afspejler langsom elimination fra ikke-plasmakompartmenter.
Virkning af antacida på absorption af ribavirin
Samtidig administration af REBETOL-kapsler med et antacidum indeholdende magnesium, aluminium og simethicon resulterede i et fald på 14 % i den gennemsnitlige AUCtf for ribavirin. Den kliniske relevans af resultater fra denne enkeltdosisundersøgelse er ukendt.
Vævsfordeling
Ribavirintransport ind i ikke-plasmakompartmenter er blevet mest omfattende undersøgt i røde blodlegemer og er blevet identificeret primært at ske via en es-type ligevægtsnukleosidtransporter. Denne type transporter er til stede på stort set alle celletyper og kan stå for det omfattende distributionsvolumen. Ribavirin binder ikke til plasmaproteiner.
Metabolisme og udskillelse
Ribavirin har to metabolismeveje: (i) en reversibel phosphoryleringsvej i kerneholdige celler; og (ii) en nedbrydningsvej, der involverer deribosylering og amidhydrolyse for at give en triazolcarboxylsyremetabolit. Ribavirin og dets triazolcarboxamid og triazolcarboxylsyremetabolitter udskilles renalt. Efter oral administration af 600 mg 14C-ribavirin blev ca. 61 % og 12 % af radioaktiviteten elimineret i henholdsvis urin og fæces i løbet af 336 timer. Uændret ribavirin udgjorde 17 % af den administrerede dosis.
Særlige Populationer
Renal dysfunktion
Ribavirins farmakokinetik blev vurderet efter administration af en enkelt oral dosis (400 mg) ribavirin til ikke-HCV-inficerede personer med varierende grader af nyreinsufficiens. Den gennemsnitlige AUCtf-værdi var tre gange højere hos forsøgspersoner med kreatininclearance-værdier mellem 10 og 30 ml/min sammenlignet med kontrolpersoner (kreatininclearance større end 90 ml/min). Hos forsøgspersoner med kreatininclearance-værdier mellem 30 og 60 ml/min var AUCtf to gange større sammenlignet med kontrolpersoner. Den øgede AUCtf ser ud til at skyldes reduktion af renal og nonrenal clearance hos disse personer. Fase 3-effektivitetsforsøg omfattede forsøgspersoner med kreatininclearance-værdier på over 50 ml/min. Ribavirins farmakokinetik med flere doser kan ikke forudsiges nøjagtigt hos patienter med nedsat nyrefunktion. Ribavirin fjernes ikke effektivt ved hæmodialyse.
Patienter med kreatininclearance mindre end 50 ml/min bør ikke behandles med REBETOL [se KONTRAINDIKATIONER ].
Leverdysfunktion
Virkningen af leverdysfunktion blev vurderet efter en enkelt oral dosis ribavirin (600 mg). De gennemsnitlige AUCtf-værdier var ikke signifikant forskellige hos forsøgspersoner med mild, moderat eller svær leverdysfunktion (Child-Pugh-klassifikation A, B eller C) sammenlignet med kontrolpersoner. Imidlertid steg de gennemsnitlige Cmax-værdier med sværhedsgraden af leverdysfunktion og var dobbelt så høj hos forsøgspersoner med svær leverdysfunktion sammenlignet med kontrolpersoner.
Ældre patienter
Farmakokinetiske evalueringer hos ældre forsøgspersoner er ikke blevet udført.
Køn
Der var ingen klinisk signifikante farmakokinetiske forskelle i et enkeltdosisforsøg med 18 mandlige og 18 kvindelige forsøgspersoner.
Pædiatriske patienter
Farmakokinetiske egenskaber med flere doser for REBETOL 200 mg kapsler og INTRON A hos pædiatriske forsøgspersoner med kronisk hepatitis C mellem 5 og 16 år er opsummeret i tabel 12. Farmakokinetikken for REBETOL 200 mg og INTRON A er ens i voksne og) pædiatriske fag.
Fuldstændige farmakokinetiske egenskaber for REBETOL oral opløsning er ikke blevet bestemt hos pædiatriske forsøgspersoner. Ribavirin Cmin-værdier var ens efter administration af REBETOL oral opløsning eller REBETOL 200 mg kapsler under 48 ugers behandling hos pædiatriske forsøgspersoner (3 til 16 år).
Et klinisk forsøg med pædiatriske forsøgspersoner med kronisk hepatitis C mellem 3 og 17 år blev udført, hvor farmakokinetikken for PegIntron og REBETOL (kapsler og oral opløsning) blev evalueret. Hos pædiatriske forsøgspersoner, der fik en kropsoverflade-justeret dosering af PegIntron på 60 mcg/m²/uge, blev det log-transformerede ratio-estimat af eksponering under doseringsintervallet forudsagt til at være 58 % [90 % CI: 141 %, 177 %] højere end observeret hos voksne, der fik 1,5 mcg/kg/uge. Farmakokinetikken af REBETOL (dosis-normaliseret) i dette forsøg svarede til dem, der blev rapporteret i et tidligere studie med REBETOL 200 mg i kombination med INTRON A hos pædiatriske forsøgspersoner og voksne.
Effekt af mad på absorption af ribavirin
Både AUCtf og Cmax steg med 70 %, når REBETOL-kapsler blev administreret sammen med et fedtrigt måltid (841 kcal, 53,8 g fedt, 31,6 g protein og 57,4 g kulhydrat) i en enkeltdosis farmakokinetisk undersøgelse [se DOSERING OG ADMINISTRATION ].
Mikrobiologi
Virkningsmekanisme
Den mekanisme, hvorved ribavirin bidrager til dets antivirale effekt i klinikken, er ikke fuldt ud forstået. Ribavirin har direkte antiviral aktivitet i vævskultur mod mange RNA-vira. Ribavirin øger mutationsfrekvensen i genomerne af flere vira, og ribavirintriphosphat hæmmer HCV-polymerase i en biokemisk reaktion.
Antiviral aktivitet i cellekultur
Den antivirale aktivitet af ribavirin i HCV-replikonet er ikke godt forstået og er ikke blevet defineret på grund af ribavirins cellulære toksicitet. Direkte antiviral aktivitet er blevet observeret i vævskulturer af andre RNA-vira. Anti-HCV-aktiviteten af interferon blev påvist i celler indeholdende selvreplikerende HCV-RNS (HCV-replikonceller) eller HCV-infektion.
Modstand
HCV-genotyper viser stor variation i deres respons på pegyleret rekombinant human interferon/ribavirinbehandling. Genetiske ændringer forbundet med det variable respons er ikke blevet identificeret.
Krydsmodstand
Der er ingen rapporteret krydsresistens mellem pegylerede/ikke-pegylerede interferoner og ribavirin.
Dyretoksikologi og -farmakologi
Langtidsundersøgelser i mus og rotter [18 til 24 måneder; doser på henholdsvis 20 til 75 og 10 til 40 mg/kg/dag (estimerede humane ækvivalente doser på henholdsvis 1,67 til 6,25 og 1,43 til 5,71 mg/kg/dag, baseret på kropsoverfladejustering for en voksen på 60 kg; ca. 0,1 til 0,4 gange den maksimale humane 24-timers dosis af ribavirin)] har vist en sammenhæng mellem kronisk ribavirin eksponering og øget forekomst af vaskulære læsioner (mikroskopiske blødninger) hos mus. Hos rotter forekom retinal degeneration hos kontrolpersoner, men forekomsten var øget hos ribavirinbehandlede rotter.
et studie, hvor rotteunger blev doseret postnatalt med ribavirin i doser på 10, 25 og 50 mg/kg/dag, forekom lægemiddelrelaterede dødsfald ved 50 mg/kg (ved plasmakoncentrationer af rotteunger under humane plasmakoncentrationer hos mennesker terapeutisk dosis) mellem undersøgelsesdage 13 og 48. Rotteunger doseret fra postnatale dag 7 til 63 udviste et mindre, dosisrelateret fald i den samlede vækst ved alle doser, hvilket efterfølgende viste sig som et lille fald i kropsvægt, krone-rump-længde, og knoglelængde. Disse virkninger viste tegn på reversibilitet, og der blev ikke observeret histopatologiske effekter på knogler. Ingen ribavirin-effekter blev observeret med hensyn til neuroadfærdsmæssig eller reproduktiv udvikling.
Kliniske Studier
Klinisk undersøgelse 1 evaluerede PegIntron monoterapi. Se PegIntron-mærkning for information om dette forsøg.
REBETOL/PegIntron kombinationsterapi
Voksne emner
Studie 2
Et randomiseret forsøg sammenlignede behandling med to PegIntron/REBETOL 200 mg regimer [PegIntron 1,5 mcg/kg subkutant én gang om ugen/REBETOL 800 mg oralt dagligt (i opdelte doser); PegIntron 1,5 mcg/kg subkutant én gang ugentlig i 4 uger derefter 0,5 mcg/kg subkutant én gang ugentlig i 44 uger/REBETOL 1000 eller 1200 mg oralt dagligt (i opdelte doser)] med INTRON A [3 mio. IE subkutant tre gange ugentlig/REBET eller REBET. 1200 mg oralt dagligt (i opdelte doser)] hos 1530 voksne med kronisk hepatitis C. Interferon-naive forsøgspersoner blev behandlet i 48 uger og fulgt i 24 uger efter behandling. Kvalificerede forsøgspersoner havde kompenseret leversygdom, påviselig HCV-RNA, forhøjet ALT og leverhistopatologi i overensstemmelse med kronisk hepatitis.
Respons på behandling blev defineret som upåviselig HCV-RNA 24 uger efter behandling (se tabel 13). Responsraten på PegIntron 1,5 mcg/kg og ribavirin 800 mg dosis var højere end responsraten på INTRON A/REBETOL (se tabel 13). Responsraten på PegIntron 1,5→0,5 mcg/kg/REBETOL 200 mg var stort set den samme som svar på INTRON A/REBETOL (data ikke vist).
Individer med viral genotype 1, uanset viral belastning, havde en lavere responsrate på PegIntron (1,5 mcg/kg)/REBETOL (800 mg) sammenlignet med forsøgspersoner med andre virale genotyper. Forsøgspersoner med både dårlige prognostiske faktorer (genotype 1 og høj viral load) havde en responsrate på 30 % (78/256) sammenlignet med en responsrate på 29 % (71/247) med INTRON A/REBETOL kombinationsbehandling.
Forsøgspersoner med lavere kropsvægt havde en tendens til at have højere bivirkningsfrekvenser [se BIVIRKNINGER og højere responsrater end forsøgspersoner med højere kropsvægt. Forskelle i responsrater mellem behandlingsarme varierede ikke væsentligt med kropsvægt.
Behandlingsresponsrater med PegIntron/REBETOL kombinationsbehandling var 49 % hos mænd og 56 % hos kvinder. Responsraterne var lavere hos afroamerikanske og latinamerikanske forsøgspersoner og højere hos asiater sammenlignet med kaukasiere. Selvom afroamerikanere havde en højere andel af dårlige prognostiske faktorer sammenlignet med kaukasiere, var antallet af ikke-kaukasiere undersøgt (11% af det samlede antal) utilstrækkeligt til at tillade meningsfulde konklusioner om forskelle i responsrater efter justering for prognostiske faktorer i dette forsøg.
Leverbiopsier blev taget før og efter behandling hos 68 % af forsøgspersonerne. Sammenlignet med baseline blev ca. to tredjedele af forsøgspersonerne i alle behandlingsgrupper observeret at have en beskeden reduktion i inflammation.
Studie 3
et stort amerikansk samfundsbaseret forsøg blev 4913 forsøgspersoner med kronisk hepatitis C randomiseret til at modtage PegIntron 1,5 mcg/kg subkutant en gang om ugen i kombination med en REBETOL 200 mg dosis på 800 til 1400 mg (vægtbaseret dosering [WBD]) eller 800 mg (flad) oralt dagligt (i opdelte doser) i 24 eller 48 uger baseret på genotype. Respons på behandling blev defineret som ikke-detekterbart HCV-RNA (baseret på et assay med en nedre detektionsgrænse på 125 IE/ml) 24 uger efter behandling.
Behandling med PegIntron 1,5 mcg/kg og REBETOL 800 til 1400 mg resulterede i et højere vedvarende virologisk respons sammenlignet med PegIntron i kombination med en flad 800 mg daglig dosis af REBETOL. Forsøgspersoner, der vejede mere end 105 kg, opnåede den største fordel med WBD, selvom en beskeden fordel også blev observeret hos forsøgspersoner, der vejede mere end 85 til 105 kg (se tabel 14). Fordelen ved WBD hos forsøgspersoner, der vejer mere end 85 kg, blev observeret med HCV genotype 1-3. Der var utilstrækkelige data tilgængelige til at nå konklusioner vedrørende andre genotyper. Brug af WBD resulterede i en øget forekomst af anæmi [se BIVIRKNINGER ].
I alt 1552 forsøgspersoner, der vejede mere end 65 kg i undersøgelse 3, havde genotype 2 eller 3 og blev randomiseret til 24 eller 48 ugers behandling. Der blev ikke observeret yderligere fordele med den længere behandlingsvarighed.
Studie 4
Et stort randomiseret forsøg sammenlignede sikkerheden og effektiviteten af behandlingen i 48 uger med to PegIntron/REBETOL 200 mg regimer [PegIntron 1,5 mcg/kg og 1 mcg/kg subkutant en gang om ugen, begge i kombination med REBETOL 800 til 1400 mg PO dagligt (i to opdelt) doser)] og Pegasys 180 mcg subkutant én gang ugentligt i kombination med Copegus 1000 til 1200 mg PO dagligt (i to opdelte doser) hos 3070 behandlingsnaive voksne med kronisk hepatitis C genotype 1. I dette forsøg, mangel på tidlig virologisk respons (ikke påviselig) HCV-RNA eller større end eller lig med 2 log10 reduktion fra baseline) ved behandling. Uge 12 var kriteriet for seponering af behandlingen. SVR blev defineret som ikke-detekterbart HCV-RNA (Roche COBAS TaqMan-assay, en nedre kvantificeringsgrænse på 27 IE/ml) 24 uger efter behandling (se tabel 15).
Samlede SVR-rater var ens blandt de tre behandlingsgrupper. Uanset behandlingsgruppe var SVR-raterne lavere hos forsøgspersoner med dårlige prognostiske faktorer. Individer med dårlige prognostiske faktorer randomiseret til PegIntron (1,5 mcg/kg)/REBETOL eller Pegasys/Copegus opnåede imidlertid højere SVR-rater sammenlignet med lignende forsøgspersoner randomiseret til PegIntron 1 mcg/kg/REBETOL. For dosen PegIntron 1,5 mcg/kg og REBETOL var SVR-raterne for forsøgspersoner med og uden følgende prognostiske faktorer som følger: skrumpelever (10 % vs. 42 %), normale ALAT-niveauer (32 % vs. 42 %), viral baseline belastning større end 600.000 IE/mL (35 % vs. 61 %), 40 år og ældre (38 % vs. 50 %) og afroamerikansk race (23 % vs. 44 %). Hos forsøgspersoner med ikke-detekterbart HCV-RNA i behandlingsuge 12, som fik PegIntron (1,5 mcg/kg)/REBETOL 200 mg, var SVR-raten 81 % (328/407).
Undersøgelse 5 - REBETOL/PegIntron kombinationsterapi ved tidligere behandlingssvigt
et ikke-sammenlignende forsøg blev 2293 forsøgspersoner med moderat til svær fibrose, som har svigtet tidligere behandling med alfa-interferon/ribavirin, genbehandlet med PegIntron, 1,5 mcg/kg subkutant, én gang ugentligt, i kombination med vægtjusteret ribavirin. Kvalificerede forsøgspersoner omfattede tidligere non-responders (personer, der var HCV-RNA-positive ved slutningen af mindst 12 ugers behandling) og tidligere recidiverende (personer, der var HCVRNA-negative ved afslutningen af minimum 12 ugers behandling og efterfølgende recidiverede efter efterbehandlingen) opfølgning). Forsøgspersoner, der var negative i uge 12, blev behandlet i 48 uger og fulgt i 24 uger efter behandlingen. Respons på behandling blev defineret som upåviselig HCV-RNA 24 uger efter behandling (målt ved hjælp af en forskningsbaseret test, detektionsgrænse 125 IE/ml). Den samlede svarprocent var 22 % (497/2293) (99 % CI: 19,5, 23,9). Forsøgspersoner med følgende karakteristika var mindre tilbøjelige til at drage fordel af genbehandling: tidligere non-respons, tidligere pegyleret interferonbehandling, signifikant brodannende fibrose eller cirrhose og genotype 1-infektion.
De genbehandlingsvedvarende virologiske responsrater efter baselinekarakteristika er opsummeret i tabel 16.
Opnåelse af et ikke-detekterbart HCV-RNA ved behandlingsuge 12 var en stærk forudsigelse for SVR. I dette forsøg opnåede 1470 (64%) forsøgspersoner ikke et upåviselig HCV-RNA i behandlingsuge 12 og blev tilbudt optagelse i langtidsbehandlingsforsøg på grund af et utilstrækkeligt behandlingsrespons. Af de 823 (36 %) forsøgspersoner, som ikke kunne påvises med HCV-RNA i behandlingsuge 12, havde de inficerede med genotype 1 en SVR på 48 % (245/507), med en række responser efter fibrose-score (F4-F2) på 39-55 %. Forsøgspersoner inficeret med genotype 2/3, som ikke kunne påvises med HCV-RNA ved behandlingsuge 12, havde en samlet SVR på 70 % (196/281), med en række responser ved fibrose-score (F4-F2) på 60-83 %. For alle genotyper var højere fibrose-scores forbundet med en nedsat sandsynlighed for at opnå SVR.
Pædiatriske emner
Tidligere ubehandlede pædiatriske forsøgspersoner i alderen 3 til 17 år med kompenseret kronisk hepatitis C og påviselig HCV-RNA blev behandlet med REBETOL 15 mg/kg per dag og PegIntron 60 mcg/m² én gang ugentligt i 24 eller 48 uger baseret på HCV-genotype og baseline viral belastning. Alle forsøgspersoner skulle følges i 24 uger efter behandlingen. I alt 107 forsøgspersoner modtog behandling, hvoraf 52 % var kvinder, 89 % var kaukasiske, og 67 % var inficeret med HCV genotype 1. Forsøgspersoner inficeret med genotype 1, 4 eller genotype 3 med HCV-RNA større end eller lig med 600.000 IE/ml modtog 48 ugers behandling, mens de inficerede med genotype 2 eller genotype 3 med HCV-RNA mindre end 600.000 IE/ml fik 24 ugers behandling. Forsøgsresultaterne er opsummeret i tabel 17.
REBETOL/INTRON En kombinationsterapi
Voksne emner
Tidligere ubehandlede emner
Voksne med kompenseret kronisk hepatitis C og påviselig HCV-RNA (vurderet af et centralt laboratorium ved hjælp af et forskningsbaseret RT-PCR-assay), som tidligere var ubehandlet med alfa-interferonterapi, blev indskrevet i to multicenter, dobbeltblindede forsøg (amerikanske og internationale) og randomiseret til at modtage REBETOL 200 mg kapsler 1200 mg/dag (1000 mg/dag for forsøgspersoner, der vejer mindre end eller lig med 75 kg) og INTRON A 3 mio. ugentligt eller INTRON A og placebo i 24 eller 48 uger efterfulgt af 24 ugers opfølgning uden for terapi. Det internationale forsøg indeholdt ikke en 24-ugers INTRON A- og placebobehandlingsarm. Det amerikanske forsøg inkluderede 912 forsøgspersoner, som ved baseline var 67 % mænd, 89 % kaukasiske med en gennemsnitlig Knodell HAI-score (I+II+III) på 7,5 og 72 % genotype 1. Det internationale forsøg, udført i Europa, Israel , Canada og Australien, indskrev 799 forsøgspersoner (65 % mænd, 95 % kaukasiske, gennemsnitlig Knodell-score 6,8 og 58 % genotype 1).
Forsøgsresultater er opsummeret i tabel 18.
Af forsøgspersoner, der ikke havde opnået HCV-RNA under detektionsgrænsen for det forskningsbaserede assay inden uge 24 af REBETOL/INTRON A-behandling, reagerede mindre end 5 % på yderligere 24 ugers kombinationsbehandling.
Blandt forsøgspersoner med HCV genotype 1 behandlet med REBETOL/INTRON A-behandling, som opnåede HCV-RNA under detektionsgrænsen for det forskningsbaserede assay med 24 uger, havde de randomiseret til 48 ugers behandling højere virologiske responser sammenlignet med dem i 24- uges behandlingsgruppe. Der blev ikke observeret stigning i responsrater for forsøgspersoner med HCV non-genotype 1 randomiseret til REBETOL/INTRON A-behandling i 48 uger sammenlignet med 24 uger.
Tilbagefaldsfag
Forsøgspersoner med kompenseret kronisk hepatitis C og påviselig HCV-RNA (vurderet af et centrallaboratorium ved hjælp af et forskningsbaseret RT-PCR-assay), som havde fået tilbagefald efter en eller to forløb med interferonbehandling (defineret som unormale serum-ALAT-niveauer) blev indskrevet i to multicenter, dobbeltblindede forsøg (amerikanske og internationale) og randomiseret til at modtage REBETOL 1200 mg/dag (1000 mg/dag for forsøgspersoner, der vejer ≤ 75 kg) og INTRON A 3 mio. ugentligt eller INTRON A og placebo i 24 uger efterfulgt af 24 ugers opfølgning uden for behandlingen. Det amerikanske forsøg omfattede 153 forsøgspersoner, som ved baseline var 67 % mænd, 92 % kaukasiske med en gennemsnitlig Knodell HAI-score (I+II+III) på 6,8 og 58 % genotype 1. Det internationale forsøg, udført i Europa, Israel , Canada og Australien, indskrev 192 forsøgspersoner (64 % mænd, 95 % kaukasiske, gennemsnitlig Knodell-score 6,6 og 56 % genotype 1). Forsøgsresultater er opsummeret i tabel 19.
Virologiske og histologiske responser var ens blandt mandlige og kvindelige forsøgspersoner i både de tidligere ubehandlede og tilbagefaldsforsøg.
Pædiatriske emner
Pædiatriske forsøgspersoner i alderen 3 til 16 år med kompenseret kronisk hepatitis C og påviselig HCV-RNA (vurderet af et centrallaboratorium ved hjælp af en forskningsbaseret RT-PCR-analyse) blev behandlet med REBETOL 15 mg/kg pr. dag og INTRON A 3 mio. m² tre gange ugentligt i 48 uger efterfulgt af 24 ugers opfølgning uden for behandlingen. I alt 118 forsøgspersoner modtog behandling, hvoraf 57 % var mænd, 80 % kaukasiske og 78 % genotype 1. Forsøgspersoner under 5 år fik REBETOL oral opløsning, og de 5 år eller ældre fik enten REBETOL oral opløsning eller kapsler.
Forsøgsresultater er opsummeret i tabel 20.
Individer med viral genotype 1, uanset viral load, havde en lavere responsrate på INTRON A/REBETOL kombinationsbehandling sammenlignet med forsøgspersoner med genotype non-1, 36 % vs. 81 %. Forsøgspersoner med både dårlige prognostiske faktorer (genotype 1 og høj viral load) havde en responsrate på 26 % (13/50).
PATIENTOPLYSNINGER
Ingen oplysninger angivet. Der henvises til ADVARSLER OG FORHOLDSREGLER afsnit.