Xeloda 500mg Capecitabine Anvendelse, bivirkninger og dosering. Pris i onlineapotek. Generisk medicin uden recept.
Hvad er Xeloda 500mg, og hvordan bruges det?
Xeloda er en receptpligtig medicin, der bruges til at behandle symptomer på kræftformer såsom tyktarmskræft, tyktarmskræft og brystkræft. Xeloda kan bruges alene eller sammen med anden medicin.
Xeloda tilhører en klasse af lægemidler kaldet Antineoplastics, Antimetabolite.
Det vides ikke, om Xeloda 500 mg er sikkert og effektivt til børn.
Hvad er de mulige bivirkninger af Xeloda 500mg?
Xeloda kan forårsage alvorlige bivirkninger, herunder:
- feber over 100,5 grader,
- kvalme,
- mistet appetiten,
- spiser meget mindre end normalt,
- opkastning (mere end én gang om 24 timer),
- svær diarré (mere end 4 gange om dagen eller om natten),
- vabler eller sår i munden,
- rødt eller hævet tandkød,
- synkebesvær,
- smerte, ømhed, rødme, hævelse, blærer eller afskallet hud på dine hænder eller fødder,
- føler dig meget tørstig eller varm,
- ude af stand til at tisse,
- kraftig svedtendens,
- varm og tør hud,
- brystsmerter eller tryk,
- ujævne hjerteslag,
- stakåndet,
- hævelse eller hurtig vægtøgning,
- smertefuld eller vanskelig vandladning,
- hævelse i dine fødder eller ankler,
- træthedsfornemmelse,
- stakåndet,
- mørk urin,
- lerfarvede afføring,
- gulfarvning af huden eller øjnene (gulsot),
- feber eller andre influenzasymptomer,
- hoste,
- hudsår,
- bleg hud,
- let blå mærker,
- usædvanlig blødning,
- føler sig svimmel,
- hurtig hjerterytme,
- ondt i halsen,
- hævelse i dit ansigt eller tunge,
- brændende i dine øjne, og
- hudsmerter, der efterfølges af et rødt eller lilla udslæt (især i ansigtet eller overkroppen) og forårsager blærer og afskalning
Få lægehjælp med det samme, hvis du har nogle af symptomerne nævnt ovenfor.
De mest almindelige bivirkninger af Xeloda 500mg omfatter:
- mavesmerter,
- forstoppelse,
- ondt i maven,
- træt følelse,
- mildt hududslæt, og
- følelsesløshed eller prikken i dine hænder eller fødder
Fortæl det til lægen, hvis du har en bivirkning, der generer dig, eller som ikke forsvinder.
Disse er ikke alle de mulige bivirkninger af Xeloda. Spørg din læge eller apotek for mere information.
Ring til din læge for lægehjælp om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
ADVARSEL
XELODA-WARFARIN INTERAKTION
XELODA 500 mg Warfarin Interaktion: Patienter, der samtidig får capecitabin og oral coumarin-derivatet antikoagulantbehandling, bør have deres antikoagulerende respons (INR eller protrombintid) overvåget hyppigt for at justere antikoagulantdosis i overensstemmelse hermed. En klinisk vigtig XELODA-Warfarin lægemiddelinteraktion blev påvist i et klinisk farmakologisk forsøg [se ADVARSLER OG FORHOLDSREGLER og DRUGSINTERAKTIONER]. Ændrede koagulationsparametre og/eller blødning, inklusive dødsfald, er blevet rapporteret hos patienter, der tager XELODA 500 mg samtidig med coumarin-derivative antikoagulantia såsom warfarin og phenprocoumon. Postmarketing-rapporter har vist klinisk signifikante stigninger i protrombintid (PT) og INR hos patienter, som var stabiliseret på antikoagulantia på det tidspunkt, hvor XELODA 500 mg blev introduceret. Disse hændelser opstod inden for flere dage og op til flere måneder efter påbegyndelse af XELODA 500 mg-behandling og i nogle få tilfælde inden for 1 måned efter ophør med XELODA. Disse hændelser forekom hos patienter med og uden levermetastaser. Alder over 60 og en diagnose af cancer disponerer selvstændigt patienter for en øget risiko for koagulopati.
BESKRIVELSE
XELODA (capecitabin) er et fluoropyrimidincarbamat med antineoplastisk aktivitet. Det er et oralt administreret systemisk prodrug af 5'-deoxy-5-fluorouridin (5'-DFUR), som omdannes til 5-fluorouracil.
Det kemiske navn for capecitabin er 5'-deoxy-5-fluor-N-[(pentyloxy)carbonyl]-cytidin og har en molekylvægt på 359,35. Capecitabin har følgende strukturformel:
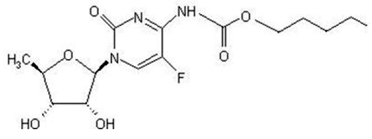
Capecitabin er et hvidt til råhvidt krystallinsk pulver med en vandopløselighed på 26 mg/ml ved 20°C.
XELODA leveres som bikonvekse, aflange filmovertrukne tabletter til oral administration. Hver lys ferskenfarvet tablet indeholder 150 mg capecitabin, og hver ferskenfarvet tablet indeholder 500 mg capecitabin. De inaktive ingredienser i XELODA omfatter: vandfri lactose, croscarmellosenatrium, hydroxypropylmethylcellulose, mikrokrystallinsk cellulose, magnesiumstearat og renset vand. Belægningen af fersken eller lys ferskenfilm indeholder hydroxypropylmethylcellulose, talkum, titaniumdioxid og syntetiske gule og røde jernoxider.
INDIKATIONER
Kolorektal cancer
- XELODA er indiceret som enkeltstof til adjuverende behandling hos patienter med Dukes' C coloncancer, som har gennemgået fuldstændig resektion af den primære tumor, når behandling med fluoropyrimidinterapi alene foretrækkes. XELODA 500 mg var non-inferior i forhold til 5fluorouracil og leucovorin (5-FU/LV) for sygdomsfri overlevelse (DFS). Læger bør overveje resultaterne af kombinationskemoterapiforsøg, som har vist forbedringer i DFS og OS, når de ordinerer enkeltstof XELODA 500 mg til adjuverende behandling af Dukes' C tyktarmskræft.
- XELODA 500 mg er indiceret som førstelinjebehandling af patienter med metastatisk kolorektalt karcinom, når behandling med fluoropyrimidin alene foretrækkes. Kombinationskemoterapi har vist en overlevelsesfordel sammenlignet med 5-FU/LV alene. En overlevelsesfordel i forhold til 5-FU/LV er ikke blevet påvist med XELODA 500 mg monoterapi. Brug af XELODA i stedet for 5FU/LV i kombinationer er ikke blevet tilstrækkeligt undersøgt for at sikre sikkerhed eller bevarelse af overlevelsesfordelen.
Brystkræft
- XELODA i kombination med docetaxel er indiceret til behandling af patienter med metastatisk brystkræft efter svigt af tidligere antracyklinholdig kemoterapi.
- XELODA 500 mg monoterapi er også indiceret til behandling af patienter med metastatisk brystkræft, der er resistente over for både paclitaxel og et antracyklinholdigt kemoterapiregime eller er resistente over for paclitaxel, og for hvem yderligere anthracyklinbehandling ikke er indiceret (f.eks. patienter, der har fået kumulative doser på 400 mg/m2 doxorubicin eller doxorubicinækvivalenter). Resistens er defineret som progressiv sygdom under behandling, med eller uden et indledende respons, eller tilbagefald inden for 6 måneder efter afsluttet behandling med et antracyklinholdigt adjuverende regime.
DOSERING OG ADMINISTRATION
Vigtige administrationsinstruktioner
XELODA tabletter skal synkes hele med vand inden for 30 minutter efter et måltid. XELODA 500mg er et cytotoksisk lægemiddel. Følg gældende særlige håndterings- og bortskaffelsesprocedurer.1 Hvis XELODA 500 mg tabletter skal skæres eller knuses, skal dette gøres af en fagmand, der er uddannet i sikker håndtering af cytotoksiske lægemidler ved hjælp af passende udstyr og sikkerhedsprocedurer. XELODA-dosis beregnes efter kropsoverfladeareal.
Standard startdosis
Monoterapi (metastatisk kolorektal cancer, adjuverende kolorektal cancer, metastatisk brystkræft)
Den anbefalede dosis af XELODA er 1250 mg/m2 indgivet oralt to gange dagligt (morgen og aften; svarende til 2500 mg/m2 samlet daglig dosis) i 2 uger efterfulgt af en 1-uges hvileperiode givet som 3-ugers cyklusser (se ).
Adjuverende behandling til patienter med Dukes' C tyktarmscancer anbefales i i alt 6 måneder [dvs. XELODA 1250 mg/m2 oralt to gange dagligt i 2 uger efterfulgt af en 1-uges hvileperiode, givet som 3-ugers cyklusser i alt af 8 cyklusser (24 uger)].
I kombination med Docetaxel (metastatisk brystkræft)
I kombination med docetaxel er den anbefalede dosis af XELODA 1250 mg/m2 to gange dagligt i 2 uger efterfulgt af en 1-uges hvileperiode, kombineret med docetaxel på 75 mg/m2 som en 1-times intravenøs infusion hver 3. uge. Præmedicinering, ifølge docetaxel-mærkningen, bør påbegyndes før docetaxel-administration til patienter, der får XELODA plus docetaxel-kombinationen. viser den samlede daglige dosis af XELODA efter kropsoverfladeareal og antallet af tabletter, der skal tages ved hver dosis.
Retningslinjer for dosishåndtering
Generel
XELODA-dosering skal muligvis individualiseres for at optimere patientbehandlingen. Patienter bør overvåges omhyggeligt for toksicitet, og doser af XELODA 500 mg bør modificeres efter behov for at imødekomme individuelle patienters tolerance over for behandling [se Kliniske Studier ]. Toksicitet på grund af administration af XELODA 500 mg kan håndteres ved symptomatisk behandling, dosisafbrydelser og justering af XELODA-dosis. Når dosis er blevet reduceret, bør den ikke øges på et senere tidspunkt. Doser af XELODA 500 mg udeladt på grund af toksicitet erstattes eller genoprettes ikke; i stedet bør patienten genoptage de planlagte behandlingscyklusser.
Dosen af phenytoin og dosis af coumarin-derivat antikoagulantia skal muligvis reduceres, når begge lægemidler administreres samtidig med XELODA [se DRUGSINTERAKTIONER ].
Monoterapi (metastatisk kolorektal cancer, adjuverende kolorektal cancer, metastatisk brystkræft)
XELODA dosismodifikationsskema som beskrevet nedenfor (se ) anbefales til behandling af bivirkninger.
I kombination med Docetaxel (metastatisk brystkræft)
Dosisændringer af XELODA 500mg for toksicitet bør foretages i henhold til ovenstående for XELODA. Ved begyndelsen af en behandlingscyklus, hvis en behandlingsforsinkelse er indiceret for enten XELODA 500 mg eller docetaxel, skal administrationen af begge midler udskydes, indtil kravene for genstart af begge lægemidler er opfyldt.
Dosisreduktionsskemaet for docetaxel, når det anvendes i kombination med XELODA til behandling af metastatisk brystkræft, er vist i tabel 3.
Justering af startdosis i særlige populationer
Nedsat nyrefunktion
Ingen justering af startdosis af XELODA 500 mg anbefales til patienter med let nedsat nyrefunktion (kreatininclearance = 51 til 80 ml/min [Cockroft og Gault, som vist nedenfor]). Hos patienter med moderat nedsat nyrefunktion (baseline kreatininclearance = 30 til 50 ml/min), en dosisreduktion til 75 % af XELODA-startdosis, når det anvendes som monoterapi eller i kombination med docetaxel (fra 1250 mg/m2 til 950 mg/m2) to gange dagligt) anbefales [se Brug i specifikke populationer og KLINISK FARMAKOLOGI ]. Efterfølgende dosisjustering anbefales som beskrevet i og (afhængigt af regimen), hvis en patient udvikler en grad 2 til 4 uønsket hændelse [se ADVARSLER OG FORHOLDSREGLER ]. Anbefalingerne for startdosisjustering for patienter med moderat nedsat nyrefunktion gælder for både XELODA 500 mg monoterapi og XELODA 500 mg i kombination med docetaxel.
Cockroft og Gault ligning:
Geriatri
Læger bør udvise forsigtighed med at overvåge virkningerne af XELODA 500 mg hos ældre. Der er utilstrækkelige data til at give en dosisanbefaling.
HVORDAN LEVERET
Doseringsformer Styrker
XELODA leveres som bikonvekse, aflange filmovertrukne tabletter til oral administration. Hver lys ferskenfarvet tablet indeholder 150 mg capecitabin, og hver ferskenfarvet tablet indeholder 500 mg capecitabin.
150 mg
Farve: Lys fersken Gravering: XELODA på den ene side og 150 på den anden 150 mg tabletter er pakket i flasker med 60 ( NDC 0004-1100-20), individuelt pakket i en karton.
500 mg
Farve: Ferskengravering: XELODA 500 mg på den ene side og 500 på den anden 500 mg tabletter er pakket i flasker med 120 ( NDC 0004-1101-50), individuelt pakket i en karton.
Opbevaring og håndtering
Opbevares ved 25°C (77°F); udflugter tilladt til 15° til 30°C (59° til 86°F). [Se USP-styret rumtemperatur]. HOLD TÆT LUKKET.
XELODA er et cytotoksisk lægemiddel. Følg relevante særlige håndterings- og bortskaffelsesprocedurer.1 Ethvert ubrugt produkt skal bortskaffes i overensstemmelse med lokale krav eller lægemiddeltilbagetagelsesprogrammer.
REFERENCER
1. "OSHA farlige stoffer." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Distribueret af: Genentech USA, Inc. Medlem af Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Revideret: maj 2021
BIVIRKNINGER
Fordi kliniske forsøg udføres under vidt forskellige forhold, kan bivirkningsrater observeret i de kliniske forsøg med et lægemiddel ikke direkte sammenlignes med hastigheder i de kliniske forsøg med et andet lægemiddel og afspejler muligvis ikke de frekvenser, der observeres i praksis.
Adjuverende tyktarmskræft
viser de bivirkninger, der forekommer hos ≥5 % af patienterne fra et fase 3-studie hos patienter med Dukes' C tyktarmskræft, som modtog mindst én dosis undersøgelsesmedicin og havde mindst én sikkerhedsvurdering. I alt 995 patienter blev behandlet med 1250 mg/m2 to gange dagligt af XELODA 500 mg administreret i 2 uger efterfulgt af en 1-uges hvileperiode, og 974 patienter fik administreret 5-FU og leucovorin (20 mg/m2 leucovorin IV efterfulgt af 425 mg/m2 IV bolus 5FU på dag 1-5 hver 28. dag). Medianbehandlingsvarigheden var 164 dage for patienter behandlet med capecitabinet og 145 dage for 5-FU/LV-behandlede patienter. I alt 112 (11 %) og 73 (7 %) capecitabin- og 5-FU/LV-behandlede patienter afbrød behandlingen på grund af bivirkninger. I alt 18 dødsfald på grund af alle årsager indtraf enten på studiet eller inden for 28 dage efter modtagelse af studielægemidlet: 8 (0,8 %) patienter randomiseret til XELODA 500 mg og 10 (1,0 %) randomiserede til 5-FU/LV.
viser grad 3/4 laboratorieabnormiteter, der forekommer hos ≥1 % af patienterne fra et fase 3-studie hos patienter med Dukes' C tyktarmskræft, som fik mindst én dosis undersøgelsesmedicin og havde mindst én sikkerhedsvurdering.
Metastatisk tyktarmskræft
Monoterapi
viser de bivirkninger, der forekommer hos ≥5 % af patienterne ved at samle de to fase 3-forsøg i førstelinjemetastatisk kolorektal cancer. I alt 596 patienter med metastatisk kolorektal cancer blev behandlet med 1250 mg/m2 to gange dagligt af XELODA administreret i 2 uger efterfulgt af en 1-uges hvileperiode, og 593 patienter fik administreret 5-FU og leukovorin i Mayo-kuren (20 mg/m2 leucovorin IV efterfulgt af 425 mg/m2 IV bolus 5-FU på dag 1-5 hver 28. dag). I den samlede kolorektale database var den gennemsnitlige behandlingsvarighed 139 dage for capecitabin-behandlede patienter og 140 dage for 5-FU/LV-behandlede patienter. I alt 78 (13%) og 63 (11%) capecitabin- og 5-FU/LV-behandlede patienter afbrød behandlingen på grund af bivirkninger/interkurrent sygdom. I alt 82 dødsfald på grund af alle årsager indtraf enten i undersøgelsen eller inden for 28 dage efter modtagelse af forsøgslægemidlet: 50 (8,4 %) patienter randomiseret til XELODA 500 mg og 32 (5,4 %) randomiserede til 5-FU/LV.
Brystkræft
I kombination med Docetaxel
Følgende data er vist for kombinationsstudiet med XELODA 500mg og docetaxel hos patienter med metastaserende brystkræft i og . I kombinationsarmen XELODA og docetaxel var behandlingen XELODA administreret oralt 1250 mg/m2 to gange dagligt som intermitterende terapi (2 ugers behandling efterfulgt af 1 uge uden behandling) i mindst 6 uger og docetaxel administreret som en 1-times intravenøs infusion kl. en dosis på 75 mg/m2 på den første dag i hver 3-ugers cyklus i mindst 6 uger. I monoterapiarmen blev docetaxel administreret som en 1-times intravenøs infusion i en dosis på 100 mg/m2 på den første dag i hver 3-ugers cyklus i mindst 6 uger. Den gennemsnitlige behandlingsvarighed var 129 dage i kombinationsarmen og 98 dage i monoterapiarmen. I alt 66 patienter (26 %) i kombinationsarmen og 49 (19 %) i monoterapiarmen trak sig ud af undersøgelsen på grund af bivirkninger. Procentdelen af patienter, der havde behov for dosisreduktion på grund af bivirkninger, var 65 % i kombinationsarmen og 36 % i monoterapiarmen. Procentdelen af patienter, der krævede behandlingsafbrydelser på grund af bivirkninger i kombinationsarmen, var 79 %. Behandlingsafbrydelser var en del af dosismodifikationsskemaet for kombinationsterapiarmen, men ikke for de docetaxel-monoterapibehandlede patienter.
Monoterapi
Følgende data er vist for undersøgelsen i stadium IV brystkræftpatienter, som fik en dosis på 1250 mg/m2 administreret to gange dagligt i 2 uger efterfulgt af en 1-uges hvileperiode. Den gennemsnitlige behandlingsvarighed var 114 dage. I alt 13 ud af 162 patienter (8%) ophørte med behandlingen på grund af bivirkninger/interkurrent sygdom.
Klinisk relevante bivirkninger hos
Klinisk relevante bivirkninger rapporteret hos
Monoterapi (metastatisk kolorektal cancer, adjuverende kolorektal cancer, metastatisk brystkræft)
Gastrointestinale: abdominal udspiling, dysfagi, proctalgi, ascites (0,1%), mavesår (0,1%), ileus (0,3%), toksisk udvidelse af tarmen, gastroenteritis (0,1%)
Hud og subkutan.: neglelidelse (0,1 %), øget svedtendens (0,1 %), lysfølsomhedsreaktion (0,1 %), sårdannelse i huden, kløe, strålingsgenkaldelsessyndrom (0,2 %)
Generel: brystsmerter (0,2%), influenzalignende sygdom, hedeture, smerter (0,1%), hæshed, irritabilitet, gangbesvær, tørst, brystmasse, kollaps, fibrose (0,1%), blødning, ødem, sedation
Neurologisk: søvnløshed, ataksi (0,5%), tremor, dysfasi, encefalopati (0,1%), unormal koordination, dysartri, bevidsthedstab (0,2%), nedsat balance
Metabolisme: øget vægt, kakeksi (0,4%), hypertriglyceridæmi (0,1%), hypokaliæmi, hypomagnesæmi
Øje: konjunktivitis
Åndedræt: hoste (0,1 %), epistaxis (0,1 %), astma (0,2 %), hæmoptyse, åndedrætsbesvær (0,1 %), dyspnø
Hjerte: takykardi (0,1%), bradykardi, atrieflimren, ventrikulære ekstrasystoler, ekstrasystoler, myokarditis (0,1%), perikardiel effusion
Infektioner: laryngitis (1,0%), bronkitis (0,2%), lungebetændelse (0,2%), bronkopneumoni (0,2%), keratoconjunctivitis, sepsis (0,3%), svampeinfektioner (inklusive candidiasis) (0,2%)
Muskuloskeletale: myalgi, knoglesmerter (0,1%), gigt (0,1%), muskelsvaghed
Blod og lymfe: leukopeni (0,2%), koagulationsforstyrrelse (0,1%), knoglemarvsdepression (0,1%), idiopatisk trombocytopeni purpura (1,0%), pancytopeni (0,1%)
Vaskulær: hypotension (0,2%), hypertension (0,1%), lymfødem (0,1%), lungeemboli (0,2%), cerebrovaskulær ulykke (0,1%)
Psykiatrisk: depression, forvirring (0,1 %)
Nyre: nedsat nyrefunktion (0,6 %)
Øre: svimmelhed
Lever og galde: leverfibrose (0,1 %), hepatitis (0,1 %), kolestatisk hepatitis (0,1 %), unormale leverfunktionsprøver
Immunsystem: lægemiddeloverfølsomhed (0,1 %)
XELODA 500mg i kombination med docetaxel (metastatisk brystkræft)
Gastrointestinale: ileus (0,4%), nekrotiserende enterocolitis (0,4%), esophageal ulcus (0,4%), hæmoragisk diarré (0,8%)
Neurologisk: ataksi (0,4%), synkope (1,2%), smagstab (0,8%), polyneuropati (0,4%), migræne (0,4%)
Hjerte: supraventrikulær takykardi (0,4 %)
Infektion: neutropen sepsis (2,4%), sepsis (0,4%), bronkopneumoni (0,4%) Blod &
Lymfe: agranulocytose (0,4%), protrombin faldt (0,4%)
Vaskulær: hypotension (1,2%), venøs flebitis og tromboflebitis (0,4%), postural hypotension (0,8%)
Nyre: nyresvigt (0,4 %)
Lever og galde: gulsot (0,4 %), unormale leverfunktionsprøver (0,4 %), leversvigt (0,4 %), hepatisk koma (0,4 %), levertoksicitet (0,4 %)
Immunsystem: overfølsomhed (1,2 %)
Postmarketing oplevelse
Følgende bivirkninger er blevet observeret efter markedsføring: angioødem, leversvigt, lacrimal duct stenose, akut nyresvigt sekundært til dehydrering inklusive fatalt udfald [se ADVARSLER OG FORHOLDSREGLER ], kutan lupus erythematosus, hornhindelidelser inklusive keratitis, toksisk leukoencefalopati, alvorlige hudreaktioner såsom Stevens-Johnsons syndrom og toksisk epidermal nekrolyse (TEN) [se ADVARSLER OG FORHOLDSREGLER ], vedvarende eller alvorligt hånd- og fodsyndrom kan i sidste ende føre til tab af fingeraftryk [se ADVARSLER OG FORHOLDSREGLER ]
I tilfælde af eksponering for knuste XELODA 500 mg tabletter er følgende bivirkninger blevet rapporteret: øjenirritation og hævelse, hududslæt, diarré, paræstesi, hovedpine, maveirritation, opkastning og kvalme.
DRUGSINTERAKTIONER
Lægemiddel-lægemiddelinteraktioner
Antikoagulanter
Ændrede koagulationsparametre og/eller blødning er blevet rapporteret hos patienter, der tager XELODA 500 mg samtidig med antikoagulantia, der er afledt af coumarin, såsom warfarin og phenprocoumon [se KASSE ADVARSEL ]. Disse hændelser opstod inden for flere dage og op til flere måneder efter påbegyndelse af XELODA 500 mg-behandling og i nogle få tilfælde inden for 1 måned efter ophør med XELODA. Disse hændelser forekom hos patienter med og uden levermetastaser. I et lægemiddelinteraktionsstudie med enkeltdosis warfarinadministration var der en signifikant stigning i den gennemsnitlige AUC for S-warfarin [se KLINISK FARMAKOLOGI ]. Den maksimale observerede INR-værdi steg med 91 %. Denne interaktion skyldes sandsynligvis en hæmning af cytochrom P450 2C9 af capecitabin og/eller dets metabolitter.
Phenytoin
Niveauet af phenytoin bør overvåges nøje hos patienter, der tager XELODA 500 mg, og phenytoindosis skal muligvis reduceres [se DOSERING OG ADMINISTRATION ]. Postmarketing-rapporter indikerer, at nogle patienter, der fik XELODA 500 mg og phenytoin, havde toksicitet forbundet med forhøjede phenytoinniveauer. Formelle lægemiddel-interaktionsstudier med phenytoin er ikke blevet udført, men interaktionsmekanismen formodes at være hæmning af CYP2C9-isoenzymet af capecitabin og/eller dets metabolitter.
Leucovorin
Koncentrationen af 5-fluorouracil øges, og dets toksicitet kan forstærkes af leucovorin. Dødsfald som følge af svær enterocolitis, diarré og dehydrering er blevet rapporteret hos ældre patienter, der fik leucovorin og fluorouracil ugentligt.
CYP2C9 substrater
Ud over warfarin er der ikke udført formelle lægemiddelinteraktionsstudier mellem XELODA og andre CYP2C9-substrater. Der skal udvises forsigtighed, når XELODA administreres sammen med CYP2C9-substrater.
Allopurinol
Samtidig brug med allopurinol kan nedsætte koncentrationen af capecitabins aktive metabolitter [se KLINISK FARMAKOLOGI ], hvilket kan nedsætte XELODA-effekten. Undgå brugen af allopurinol under behandling med XELODA.
Lægemiddel-fødevareinteraktion
Mad blev vist at reducere både hastigheden og omfanget af absorption af capecitabin [se KLINISK FARMAKOLOGI ]. I alle kliniske forsøg blev patienterne instrueret i at administrere XELODA 500 mg inden for 30 minutter efter et måltid. Det anbefales, at XELODA administreres sammen med mad [se DOSERING OG ADMINISTRATION ].
ADVARSLER
Inkluderet som en del af "FORHOLDSREGLER" Afsnit
FORHOLDSREGLER
Koagulopati
Patienter, der samtidig får capecitabin og oral coumarin-derivatet antikoagulantbehandling, bør have deres antikoagulerende respons (INR eller protrombintid) overvåget nøje med stor hyppighed, og antikoagulantdosen bør justeres i overensstemmelse hermed [se KASSE ADVARSEL og DRUGSINTERAKTIONER ].
Diarré
XELODA 500mg kan fremkalde diarré, nogle gange alvorlig. Patienter med svær diarré bør overvåges nøje og gives væske- og elektrolytterstatning, hvis de bliver dehydrerede. Hos 875 patienter med enten metastatisk brystkræft eller kolorektal cancer, som modtog XELODA monoterapi, var mediantiden til første forekomst af grad 2 til 4 diarré 34 dage (fra 1 til 369 dage). Medianvarigheden af grad 3 til 4 diarré var 5 dage. National Cancer Institute of Canada (NCIC) grad 2 diarré er defineret som en stigning på 4 til 6 afføring/dag eller natlig afføring, grad 3 diarré som en stigning på 7 til 9 afføring/dag eller inkontinens og malabsorption, og grad 4 diarré som en stigning på ≥10 afføring/dag eller kraftig blodig diarré eller behov for parenteral støtte. Hvis der opstår grad 2, 3 eller 4 diarré, skal administrationen af XELODA straks afbrydes, indtil diarréen forsvinder eller falder i intensitet til grad 1 [se DOSERING OG ADMINISTRATION ]. Standard antidiarrébehandlinger (f.eks. loperamid) anbefales.
Nekrotiserende enterocolitis (tyflitis) er blevet rapporteret.
Kardiotoksicitet
Kardiotoksiciteten observeret med XELODA 500 mg inkluderer myokardieinfarkt/iskæmi, angina, dysrytmier, hjertestop, hjertesvigt, pludselig død, elektrokardiografiske ændringer og kardiomyopati. Disse bivirkninger kan være mere almindelige hos patienter med en tidligere koronararteriesygdom.
Dihydropyrimidin-dehydrogenase-mangel
Baseret på rapporter efter markedsføring har patienter med visse homozygote eller visse sammensatte heterozygote mutationer i DPD-genet, der resulterer i fuldstændig eller næsten fuldstændig fravær af DPD-aktivitet, øget risiko for akut tidlig indtræden af toksicitet og alvorlige, livstruende eller dødelige bivirkninger reaktioner forårsaget af XELODA (f.eks. mucositis, diarré, neutropeni og neurotoksicitet). Patienter med delvis DPD-aktivitet kan også have øget risiko for alvorlige, livstruende eller fatale bivirkninger forårsaget af XELODA.
Tilbageholde eller seponere permanent XELODA 500 mg baseret på klinisk vurdering af indtræden, varighed og sværhedsgrad af de observerede toksiciteter hos patienter med tegn på akut tidligt indsættende eller usædvanligt alvorlig toksicitet, som kan indikere næsten fuldstændig eller fuldstændig fravær af DPD-aktivitet. Ingen XELODA-dosis har vist sig at være sikker for patienter med fuldstændig fravær af DPD-aktivitet. Der er utilstrækkelige data til at anbefale en specifik dosis til patienter med delvis DPD-aktivitet målt ved en specifik test.
Dehydrering og nyresvigt
Dehydrering er blevet observeret og kan forårsage akut nyresvigt, som kan være fatalt. Patienter med allerede eksisterende kompromitteret nyrefunktion, eller som samtidig får XELODA med kendte nefrotoksiske midler, har højere risiko. Patienter med anoreksi, asteni, kvalme, opkastning eller diarré kan hurtigt blive dehydreret. Overvåg patienter, når XELODA 500 mg administreres for at forhindre og korrigere dehydrering ved debut. Hvis der opstår grad 2 (eller højere) dehydrering, skal XELODA-behandlingen straks afbrydes og dehydreringen korrigeres. Behandlingen bør ikke genoptages, før patienten er rehydreret, og eventuelle udløsende årsager er blevet korrigeret eller kontrolleret. Dosisændringer bør foretages for den udløsende uønskede hændelse efter behov [se DOSERING OG ADMINISTRATION ].
Patienter med moderat nedsat nyrefunktion ved baseline kræver dosisreduktion [se DOSERING OG ADMINISTRATION ]. Patienter med let og moderat nedsat nyrefunktion ved baseline bør overvåges nøje for bivirkninger. Hurtig afbrydelse af behandlingen med efterfølgende dosisjusteringer anbefales, hvis en patient udvikler en grad 2 til 4 uønsket hændelse som beskrevet i [se DOSERING OG ADMINISTRATION , Brug i specifikke populationer , og KLINISK FARMAKOLOGI ].
Embryo-føtal toksicitet
Baseret på resultater fra dyrereproduktionsundersøgelser og dets virkningsmekanisme, kan XELODA forårsage fosterskader, når det gives til en gravid kvinde [se KLINISK FARMAKOLOGI ]. Begrænsede tilgængelige data er ikke tilstrækkelige til at informere brugen af XELODA 500 mg til gravide kvinder. I reproduktionsstudier på dyr forårsagede administration af capecitabin til drægtige dyr i perioden med organogenese embryodødelighed og teratogenicitet hos mus og embryodødelighed hos aber ved henholdsvis 0,2 og 0,6 gange eksponeringen (AUC) hos patienter, der fik den anbefalede dosis [se hhv. Brug i specifikke populationer ]. Oplys gravide kvinder om den potentielle risiko for et foster. Rådgiv kvinder med reproduktionspotentiale til at bruge effektiv prævention under behandlingen og i 6 måneder efter den sidste dosis XELODA [se Brug i specifikke populationer ].
Mukokutan og dermatologisk toksicitet
Alvorlige mukokutane reaktioner, nogle med dødelig udgang, såsom Stevens-Johnsons syndrom og toksisk epidermal nekrolyse (TEN) kan forekomme hos patienter behandlet med XELODA [se BIVIRKNINGER ]. XELODA 500 mg bør seponeres permanent hos patienter, som oplever en alvorlig mukokutan reaktion, der muligvis kan tilskrives XELODA-behandling.
Hånd- og fodsyndrom (palmar-plantar erythrodysestesi eller kemoterapi-induceret acral erytem) er en kutan toksicitet. Mediantid til debut var 79 dage (interval fra 11 til 360 dage) med en sværhedsgrad på grad 1 til 3 for patienter, der fik XELODA 500 mg monoterapi i metastaserende omgivelser. Grad 1 er karakteriseret ved en eller flere af følgende: følelsesløshed, dysæstesi/paræstesi, snurren, smertefri hævelse eller erytem i hænder og/eller fødder og/eller ubehag, som ikke forstyrrer normale aktiviteter. Grad 2 hånd- og fodsyndrom er defineret som smertefuldt erytem og hævelse af hænder og/eller fødder og/eller ubehag, der påvirker patientens daglige aktiviteter. Grad 3 hånd-og-fod syndrom er defineret som fugtig afskalning, sårdannelse, blæredannelse eller stærke smerter i hænder og/eller fødder og/eller alvorligt ubehag, der medfører, at patienten ikke kan arbejde eller udføre daglige aktiviteter. Vedvarende eller alvorligt hånd- og fodsyndrom (grad 2 og derover) kan i sidste ende føre til tab af fingeraftryk, hvilket kan påvirke patientens identifikation. Hvis grad 2 eller 3 hånd- og fodsyndrom opstår, bør administrationen af XELODA afbrydes, indtil hændelsen forsvinder eller falder i intensitet til grad 1. Efter grad 3 hånd- og fodsyndrom bør efterfølgende doser af XELODA 500 mg nedsættes [ se DOSERING OG ADMINISTRATION ].
Hyperbilirubinæmi
Hos 875 patienter med enten metastaserende brystkræft eller kolorektal cancer, som fik mindst én dosis XELODA 1250 mg/m2 to gange dagligt som monoterapi i 2 uger efterfulgt af en 1-uges hvileperiode, forekom grad 3 (1,5-3 x ULN) hyperbilirubinæmi i 15,2 % (n=133) af patienterne og grad 4 (>3 x ULN) hyperbilirubinæmi forekom hos 3,9 % (n=34) af patienterne. Ud af 566 patienter, som havde levermetastaser ved baseline og 309 patienter uden levermetastaser ved baseline, forekom grad 3 eller 4 hyperbilirubinæmi hos henholdsvis 22,8 % og 12,3 %. Af de 167 patienter med grad 3 eller 4 hyperbilirubinæmi havde 18,6 % (n=31) også postbaseline-stigninger (grad 1 til 4, uden forhøjelser ved baseline) i alkalisk fosfatase og 27,5 % (n=46) havde postbaseline-stigninger i transaminaser kl. når som helst (ikke nødvendigvis samtidig). Størstedelen af disse patienter, 64,5 % (n=20) og 71,7 % (n=33), havde levermetastaser ved baseline. Derudover havde 57,5 % (n=96) og 35,3 % (n=59) af de 167 patienter forhøjelser (grad 1 til 4) ved både præbaseline og postbaseline i henholdsvis alkalisk fosfatase eller transaminaser. Kun 7,8 % (n=13) og 3,0 % (n=5) havde grad 3 eller 4 forhøjelser i alkalisk fosfatase eller transaminaser.
Hos de 596 patienter, der blev behandlet med XELODA 500 mg som førstelinjebehandling for metastatisk kolorektal cancer, svarede forekomsten af grad 3 eller 4 hyperbilirubinæmi til den samlede sikkerhedsdatabase for kliniske forsøg med XELODA 500 mg monoterapi. Mediantiden til indtræden for grad 3 eller 4 hyperbilirubinæmi i kolorektal cancerpopulationen var 64 dage, og median total bilirubin steg fra 8 μm/L ved baseline til 13 μm/L under behandling med XELODA. Af de 136 kolorektal cancerpatienter med grad 3 eller 4 hyperbilirubinæmi havde 49 patienter grad 3 eller 4 hyperbilirubinæmi som deres sidst målte værdi, hvoraf 46 havde levermetastaser ved baseline.
Hos 251 patienter med metastatisk brystkræft, som fik en kombination af XELODA 500 mg og docetaxel, forekom grad 3 (1,5 til 3 x ULN) hyperbilirubinæmi hos 7 % (n=17) og grad 4 (>3 x ULN) hyperbilirubinæmi forekom hos 2 % (n=5).
Hvis lægemiddelrelaterede grad 3 til 4 stigninger i bilirubin forekommer, skal administrationen af XELODA 500 mg straks afbrydes, indtil hyperbilirubinæmien falder til ≤3,0 X ULN [se anbefalede dosisændringer under DOSERING OG ADMINISTRATION ].
Hæmatologisk
Hos 875 patienter med enten metastaserende brystkræft eller kolorektal cancer, som fik en dosis på 1250 mg/m2 administreret to gange dagligt som monoterapi i 2 uger efterfulgt af en 1-uges hvileperiode, havde 3,2 %, 1,7 % og 2,4 % af patienterne grad 3 eller 4 henholdsvis neutropeni, trombocytopeni eller fald i hæmoglobin. Hos 251 patienter med metastatisk brystkræft, som fik en dosis XELODA i kombination med docetaxel, havde 68 % grad 3 eller 4 neutropeni, 2,8 % havde grad 3 eller 4 trombocytopeni, og 9,6 % havde grad 3 eller 4 anæmi.
Patienter med baseline neutrofiltal på
Geriatriske patienter
Patienter ≥80 år kan opleve en større forekomst af grad 3 eller 4 bivirkninger. Hos 875 patienter med enten metastatisk brystkræft eller kolorektal cancer, som fik XELODA 500 mg monoterapi, oplevede 62 % af de 21 patienter ≥80 år behandlet med XELODA 500 mg en behandlingsrelateret grad 3 eller 4 bivirkning: diarré hos 6 (28,6 %) , kvalme hos 3 (14,3 %), hånd- og fodsyndrom hos 3 (14,3 %) og opkastning hos 2 (9,5 %) patienter. Blandt de 10 patienter på 70 år og derover (ingen patienter var >80 år) behandlet med XELODA 500 mg i kombination med docetaxel, oplevede 30 % (3 ud af 10) af patienterne grad 3 eller 4 diarré og stomatitis, og 40 % (4 ud af 10) oplevede grad 3 hånd- og fodsyndrom.
Blandt de 67 patienter ≥60 år, der får XELODA i kombination med docetaxel, forekomsten af grad 3 eller 4 behandlingsrelaterede bivirkninger, behandlingsrelaterede alvorlige bivirkninger, seponeringer på grund af bivirkninger, behandlingsafbrydelser på grund af bivirkninger og behandling seponeringer inden for de første to behandlingscyklusser var højere end i patientgruppen
Hos 995 patienter, der fik XELODA som adjuverende behandling for Dukes' C tyktarmskræft efter resektion af den primære tumor, oplevede 41 % af de 398 patienter ≥65 år behandlet med XELODA en behandlingsrelateret grad 3 eller 4 bivirkning: hånd- og -fodsyndrom hos 75 (18,8%), diarré hos 52 (13,1%), stomatitis hos 12 (3,0%), neutropeni/granulocytopeni hos 11 (2,8%), opkastning hos 6 (1,5%) og kvalme hos 5 (1,3%) %) patienter. Hos patienter ≥65 år (alle randomiseret population; capecitabin 188 patienter, 5-FU/LV 208 patienter) behandlet for Dukes' C tyktarmskræft efter resektion af den primære tumor, er hazard ratios for sygdomsfri overlevelse og samlet overlevelse for XELODA sammenlignet med 5-FU/LV var henholdsvis 1,01 (95 % CI 0,80 – 1,27) og 1,04 (95 % CI 0,79 – 1,37).
Leverinsufficiens
Patienter med mild til moderat leverdysfunktion på grund af levermetastaser bør overvåges nøje, når XELODA administreres. Virkningen af alvorlig leverdysfunktion på dispositionen af XELODA er ikke kendt [se Brug i specifikke populationer og KLINISK FARMAKOLOGI ].
Kombination med andre lægemidler
Brug af XELODA i kombination med irinotecan er ikke blevet tilstrækkeligt undersøgt.
Patientrådgivningsinformation
Råd patienten til at læse den FDA-godkendte patientmærkning ( PATIENTOPLYSNINGER ).
Diarré
Informer patienter, der oplever grad 2 diarré (en stigning på 4 til 6 afføringer/dag eller natlig afføring) eller mere eller oplever svær blodig diarré med svære mavesmerter og feber, om at stoppe med at tage XELODA. Rådgive patienter om brugen af antidiarrébehandlinger (f.eks. loperamid) til at håndtere diarré [se ADVARSLER OG FORHOLDSREGLER ].
Kardiotoksicitet
Rådgiv patienterne om risikoen for kardiotoksicitet og omgående at kontakte deres læge eller at gå til en skadestue for nyopstået brystsmerter, åndenød, svimmelhed eller svimmelhed [se ADVARSLER OG FORHOLDSREGLER ].
Dihydropyrimidin-dehydrogenase-mangel
Råd patienter til at underrette deres læge, hvis de har en kendt DPD-mangel. Rådgiv patienter, hvis de har fuldstændig eller næsten fuldstændig fravær af DPD-aktivitet, de har en øget risiko for akut tidlig indtræden af toksicitet og alvorlige, livstruende eller fatale bivirkninger forårsaget af XELODA (f.eks. slimhindebetændelse, diarré, neutropeni og neurotoksicitet) [se ADVARSLER OG FORHOLDSREGLER ].
Dehydrering og nyresvigt
Instruer patienter, der oplever grad 2 eller højere dehydrering (IV væsker angivet ADVARSLER OG FORHOLDSREGLER ].
Vigtige administrationsinstruktioner
Råd patienterne til at sluge XELODA 500 mg tabletter hele med vand inden for 30 minutter efter et måltid. Råd til patienter og plejepersonale om ikke at knuse eller skære XELODA-tabletter. Rådgiv patienterne, hvis de ikke kan sluge XELODA-tabletter hele, at informere deres læge [se DOSERING OG ADMINISTRATION ].
Overfølsomhed og angioødem
Rådgiv patienterne om, at XELODA kan forårsage alvorlige overfølsomhedsreaktioner og angioødem. Rådgiv patienter, der har kendt overfølsomhed over for capecitabin eller 5-fluorouracil, om at informere deres læge [se KONTRAINDIKATIONER ]. Instruer patienter, der udvikler overfølsomhedsreaktioner eller slimhindesymptomer (f.eks. nældefeber, udslæt, erytem, kløe eller hævelse af ansigt, læber, tunge eller svælg, som gør det svært at synke eller trække vejret), om at stoppe med at tage XELODA og straks kontakte deres læge. eller at gå på skadestuen. [se BIVIRKNINGER ].
Kvalme
Instruer patienter, der oplever grad 2-kvalme (fødevareindtagelse betydeligt reduceret, men i stand til at spise intermitterende) eller mere om at stoppe med at tage XELODA 500 mg øjeblikkeligt og kontakte deres læge for håndtering af kvalme [se BIVIRKNINGER ].

Opkastning
Instruer patienter, der oplever grad 2 opkastning (2 til 5 episoder i en 24-timers periode) eller mere om at stoppe med at tage XELODA øjeblikkeligt og kontakte deres læge for at få styr på opkastning [se BIVIRKNINGER ].
Hånd-og-fod syndrom
Instruer patienter, der oplever grad 2 hånd- og fodsyndrom (smertefuldt erytem og hævelse af hænder og/eller fødder og/eller ubehag, der påvirker patienternes daglige aktiviteter) eller derover om at stoppe med at tage XELODA 500 mg øjeblikkeligt og kontakte deres læge . Informer patienterne om, at påbegyndelse af symptomatisk behandling anbefales, og hånd- og fodsyndrom kan føre til tab af fingeraftryk, hvilket kan påvirke personlig identifikation [se BIVIRKNINGER ].
Stomatitis
Informer patienter, der oplever grad 2 stomatitis (smertefuldt erytem, ødem eller sår i mund eller tunge, men i stand til at spise) eller derover om at stoppe med at tage XELODA øjeblikkeligt og kontakte deres læge [se BIVIRKNINGER ].
Feber og neutropeni
Informer patienter, der udvikler feber på 100,5°F eller mere eller andre tegn på potentiel infektion, om at kontakte deres læge [se BIVIRKNINGER ].
Embryo-føtal toksicitet
Informer kvinder om reproduktionspotentiale om den potentielle risiko for et foster og om at bruge effektiv prævention under behandling med XELODA 500 mg og i 6 måneder efter den sidste dosis. Råd kvinder til at informere deres sundhedsplejerske om en kendt eller formodet graviditet [se ADVARSLER OG FORHOLDSREGLER , Brug i specifikke populationer ].
Rådgiv mandlige patienter med kvindelige partnere med reproduktionspotentiale om at bruge effektiv prævention under behandling med XELODA 500 mg og i 3 måneder efter den sidste dosis [se Brug i specifikke populationer ].
Amning
Adviser kvinder om ikke at amme under behandling med XELODA 500 mg og i 2 uger efter den sidste dosis [se Brug i specifikke populationer ].
Ikke-klinisk toksikologi
Karcinogenese, mutagenese, svækkelse af fertilitet
Der er ikke udført tilstrækkelige undersøgelser af capecitabins karcinogene potentiale. Capecitabin var ikke mutagent in vitro for bakterier (Ames-test) eller pattedyrceller (kinesisk hamster V79/HPRT-genmutationsanalyse). Capecitabin var klastogent in vitro for humane perifere blodlymfocytter, men ikke klastogent in vivo for museknoglemarv (mikronukleustest). Fluorouracil forårsager mutationer i bakterier og gær. Fluorouracil forårsager også kromosomale abnormiteter i musens mikronukleustest in vivo.
undersøgelser af fertilitet og generel reproduktionsevne hos hunmus forstyrrede orale capecitabin-doser på 760 mg/kg/dag (ca. 2300 mg/m2/dag) brunsten og forårsagede følgelig et fald i fertiliteten. Hos mus, der blev gravide, overlevede ingen fostre denne dosis. Forstyrrelsen i brunst var reversibel. Hos mænd forårsagede denne dosis degenerative ændringer i testiklerne, herunder fald i antallet af spermatocytter og spermatider. I separate farmakokinetiske undersøgelser producerede denne dosis hos mus 5'-DFUR AUC-værdier omkring 0,7 gange de tilsvarende værdier hos patienter, der fik den anbefalede daglige dosis.
Brug i specifikke populationer
Graviditet
Risikooversigt
Baseret på resultater i reproduktionsstudier på dyr og dets virkningsmekanisme, kan XELODA 500 mg forårsage fosterskader, når det administreres til en gravid kvinde [se KLINISK FARMAKOLOGI ]. Begrænsede tilgængelige humane data er ikke tilstrækkelige til at informere om den lægemiddelrelaterede risiko under graviditet. I dyrereproduktionsundersøgelser forårsagede administration af capecitabin til drægtige dyr i perioden med organogenese embryodødelighed og teratogenicitet hos mus og embryodødelighed hos aber ved henholdsvis 0,2 og 0,6 gange eksponeringen (AUC) hos patienter, der fik den anbefalede dosis [se hhv. Data ]. Oplys gravide kvinder om den potentielle risiko for et foster.
Den estimerede baggrundsrisiko for alvorlige fødselsdefekter og abort for den angivne population er ukendt. I den generelle befolkning i USA er den estimerede baggrundsrisiko for alvorlige fødselsdefekter og abort i klinisk anerkendte graviditeter henholdsvis 2-4 % og 15-20 %.
Data
Dyredata
Oral administration af capecitabin til gravide mus i løbet af organogeneseperioden ved en dosis på 198 mg/kg/dag forårsagede misdannelser og embryodødelighed. I separate farmakokinetiske undersøgelser frembragte denne dosis hos mus 5'-DFUR AUC-værdier, der var ca. 0,2 gange AUC-værdierne hos patienter, der fik den anbefalede daglige dosis. Misdannelser hos mus omfattede ganespalte, anophthalmia, mikroftalmi, oligodaktyli, polydaktyli, syndaktyli, kinky hale og udvidelse af cerebrale ventrikler. Oral administration af capecitabin til gravide aber i løbet af organogeneseperioden ved en dosis på 90 mg/kg/dag forårsagede føtal dødelighed. Denne dosis gav 5'-DFUR AUC-værdier, der var ca. 0,6 gange AUC-værdierne hos patienter, der fik den anbefalede daglige dosis.
Amning
Risikooversigt
Der er ingen information om tilstedeværelsen af capecitabin i modermælk eller om dets virkning på mælkeproduktionen eller det ammede spædbarn. Capecitabin-metabolitter var til stede i mælken fra diegivende mus [se Data ]. På grund af potentialet for alvorlige bivirkninger fra capecitabineksponering hos ammede spædbørn, rådgive kvinder om ikke at amme under behandling med XELODA 500 mg og i 2 uger efter den sidste dosis.
Data
Diegivende mus, der fik en enkelt oral dosis capecitabin, udskilte betydelige mængder capecitabin-metabolitter i mælken.
Hunner og mænd med reproduktionspotentiale
Graviditetstest
Graviditetstest anbefales til kvinder med reproduktionspotentiale før påbegyndelse af XELODA.
Svangerskabsforebyggelse
Kvinder
XELODA 500mg kan forårsage fosterskader, når det administreres til en gravid kvinde [se Graviditet ]. Rådgiv kvinder med reproduktionspotentiale til at bruge effektiv prævention under behandlingen og i 6 måneder efter den sidste dosis af XELODA.
Hanner
Baseret på fund af genetisk toksicitet, rådgiv mandlige patienter med kvindelige partnere med reproduktionspotentiale om at bruge effektiv prævention under behandlingen og i 3 måneder efter den sidste dosis af XELODA [se Ikke-klinisk toksikologi ].
Infertilitet
Baseret på dyreforsøg kan XELODA svække fertiliteten hos kvinder og mænd med reproduktionspotentiale [se Ikke-klinisk toksikologi ].
Pædiatrisk brug
Sikkerheden og effektiviteten af XELODA hos pædiatriske patienter er ikke fastlagt. Der blev ikke påvist nogen klinisk fordel i to enkeltarmsforsøg med pædiatriske patienter med nydiagnosticerede hjernestammegliomer og højgradige gliomer. I begge forsøg modtog pædiatriske patienter en pædiatrisk undersøgelsesformulering af capecitabin samtidig med og efter afslutning af strålebehandling (samlet dosis på 5580 cGy i 180 cGy-fraktioner). Den relative biotilgængelighed af undersøgelsesformuleringen til XELODA 500 mg var ens.
Det første forsøg blev udført på 22 pædiatriske patienter (medianalder 8 år, interval 5-17 år) med nyligt diagnosticerede ikke-dissaminerede iboende diffuse hjernestammegliomer og højgradige gliomer. I den dosisfindende del af forsøget fik patienterne capecitabin med samtidig strålebehandling i doser fra 500 mg/m2 til 850 mg/m2 hver 12. time i op til 9 uger. Efter 2 ugers pause fik patienterne 1250 mg/m2 capecitabin hver 12. time på dag 1-14 i en 21 dages cyklus i op til 3 cyklusser. Den maksimalt tolererede dosis (MTD) af capecitabin administreret samtidig med strålebehandling var 650 mg/m2 hver 12. time. De vigtigste dosisbegrænsende toksiciteter var palmar-plantar erythrodysestesi og forhøjelse af alaninaminotransferase (ALAT).
Det andet forsøg blev udført i 34 yderligere pædiatriske patienter med nyligt diagnosticerede ikke-dissminerede iboende diffuse hjernestammegliomer (medianalder 7 år, interval 3-16 år) og 10 pædiatriske patienter, som modtog MTD af capecitabin i dosisfindingsstudiet og mødte berettigelseskriterierne for dette forsøg. Alle patienter fik 650 mg/m2 capecitabin hver 12. time med samtidig strålebehandling i op til 9 uger. Efter 2 ugers pause fik patienterne 1250 mg/m2 capecitabin hver 12. time på dag 1-14 i en 21-dages cyklus i op til 3 cyklusser.
Der var ingen forbedring i et års progressionsfri overlevelsesrate og et års samlet overlevelsesrate hos pædiatriske patienter med nydiagnosticerede iboende hjernestammegliomer, som fik capecitabin i forhold til en tilsvarende population af pædiatriske patienter, som deltog i andre kliniske forsøg.
Bivirkningsprofilen for capecitabin var i overensstemmelse med den kendte bivirkningsprofil hos voksne, med undtagelse af laboratorieabnormiteter, som forekom mere almindeligt hos pædiatriske patienter. De hyppigst rapporterede laboratorieabnormiteter (incidens pr. patient ≥40%) var øget ALAT (75%), lymfocytopeni (73%), leukopeni (73%), hypokaliæmi (68%), trombocytopeni (57%), hypoalbuminæmi (55). %), neutropeni (50 %), lav hæmatokrit (50 %), hypocalcæmi (48 %), hypofosfatæmi (45 %) og hyponatriæmi (45 %).
Geriatrisk brug
Læger bør være særlig opmærksomme på at overvåge bivirkningerne af XELODA 500mg hos ældre [se ADVARSLER OG FORHOLDSREGLER ].
Leverinsufficiens
Udvis forsigtighed, når patienter med mild til moderat leverdysfunktion på grund af levermetastaser behandles med XELODA. Virkningen af alvorlig leverdysfunktion på XELODA kendes ikke [se ADVARSLER OG FORHOLDSREGLER og KLINISK FARMAKOLOGI ].
Nyreinsufficiens
Patienter med moderat (kreatininclearance = 30 til 50 ml/min) og svær (kreatininclearance KONTRAINDIKATIONER , ADVARSLER OG FORHOLDSREGLER , DOSERING OG ADMINISTRATION , og KLINISK FARMAKOLOGI ].
OVERDOSIS
Symptomerne på akut overdosis vil omfatte kvalme, opkastning, diarré, mave-tarm-irritation og blødning og knoglemarvsdepression. Medicinsk behandling af overdosering bør omfatte sædvanlige understøttende medicinske indgreb, der sigter mod at korrigere de aktuelle kliniske manifestationer. Selvom der ikke er rapporteret klinisk erfaring med brug af dialyse som behandling af XELODA 500 mg overdosis, kan dialyse være en fordel ved at reducere cirkulerende koncentrationer af 5'-DFUR, en lavmolekylær metabolit af moderstoffet.
Enkeltdoser af XELODA 500 mg var ikke dødelige for mus, rotter og aber ved doser op til 2000 mg/kg (2,4, 4,8 og 9,6 gange den anbefalede humane daglige dosis på mg/m2-basis).
KONTRAINDIKATIONER
Svært nedsat nyrefunktion
XELODA er kontraindiceret til patienter med svært nedsat nyrefunktion (kreatininclearance under 30 ml/min [Cockroft og Gault]) [se Brug i specifikke populationer og KLINISK FARMAKOLOGI ].
Overfølsomhed
XELODA 500mg er kontraindiceret til patienter med kendt overfølsomhed over for capecitabin eller nogen af dets komponenter. XELODA er kontraindiceret til patienter, som har en kendt overfølsomhed over for 5fluorouracil.
KLINISK FARMAKOLOGI
Handlingsmekanisme
Enzymer omdanner capecitabin til 5-fluorouracil (5-FU) in vivo. Både normale celler og tumorceller metaboliserer 5-FU til 5-fluor-2'-deoxyuridinmonofosfat (FdUMP) og 5-fluoruridintrifosfat (FUTP). Disse metabolitter forårsager celleskade ved to forskellige mekanismer. Først binder FdUMP og folatcofaktoren, N5-10-methylentetrahydrofolat, til thymidylatsyntase (TS) for at danne et kovalent bundet ternært kompleks. Denne binding hæmmer dannelsen af thymidylat fra 2'-deoxyuridylat. Thymidylat er den nødvendige forløber for thymidintriphosphat, som er essentiel for syntesen af DNA, således at en mangel på denne forbindelse kan hæmme celledeling. For det andet kan nukleare transkriptionelle enzymer fejlagtigt inkorporere FUTP i stedet for uridintrifosfat (UTP) under syntesen af RNA. Denne metaboliske fejl kan forstyrre RNA-behandling og proteinsyntese.
Farmakokinetik
Absorption
Efter oral administration af 1255 mg/m2 BID til cancerpatienter nåede capecitabin maksimale blodniveauer i løbet af ca. 1,5 timer (Tmax), med maksimale 5-FU-niveauer lidt senere, efter 2 timer. Mad reducerede både hastigheden og omfanget af absorption af capecitabin med gennemsnitlig Cmax og AUC0-∞ faldt med henholdsvis 60 % og 35 %. Cmax og AUC0-∞ for 5-FU blev også reduceret ved mad med henholdsvis 43 % og 21 %. Mad forsinket Tmax for både forældre og 5-FU med 1,5 time [se ADVARSLER OG FORHOLDSREGLER , DOSERING OG ADMINISTRATION , og Lægemiddel-fødevareinteraktion ].
Farmakokinetikken af XELODA 500 mg og dets metabolitter er blevet evalueret hos omkring 200 cancerpatienter over et dosisområde på 500 til 3500 mg/m2/dag. Over dette interval var farmakokinetikken af XELODA og dets metabolit, 5'-DFCR, dosisproportional og ændrede sig ikke over tid. Stigningen i AUC'erne for 5'-DFUR og 5-FU var imidlertid større end proportional med stigningen i dosis, og AUC'en for 5-FU var 34 % højere på dag 14 end på dag 1. Variabiliteten mellem patienter i Cmax og AUC for 5-FU var større end 85 %.
Fordeling
Plasmaproteinbinding af capecitabin og dets metabolitter er mindre end 60 % og er ikke koncentrationsafhængig. Capecitabin var primært bundet til humant albumin (ca. 35%). XELODA 500mg har et lavt potentiale for farmakokinetiske interaktioner relateret til plasmaproteinbinding.
Bioaktivering og metabolisme
Capecitabin metaboliseres i vid udstrækning enzymatisk til 5-FU. I leveren hydrolyserer en 60 kDa carboxylesterase meget af forbindelsen til 5'-deoxy-5-fluorocytidin (5'-DFCR). Cytidindeaminase, et enzym, der findes i de fleste væv, inklusive tumorer, omdanner efterfølgende 5'-DFCR til 5'-DFUR. Enzymet, thymidinphosphorylase (dThdPase), hydrolyserer derefter 5'DFUR til det aktive lægemiddel 5-FU. Mange væv i hele kroppen udtrykker thymidinfosphorylase. Nogle humane karcinomer udtrykker dette enzym i højere koncentrationer end omgivende normale væv. Efter oral administration af XELODA 7 dage før operation hos patienter med kolorektal cancer var medianforholdet mellem 5-FU-koncentration i kolorektale tumorer og tilstødende væv 2,9 (interval fra 0,9 til 8,0). Disse forhold er ikke blevet evalueret hos brystkræftpatienter eller sammenlignet med 5-FU-infusion.
Metabolisk vej af capecitabin til 5-FU
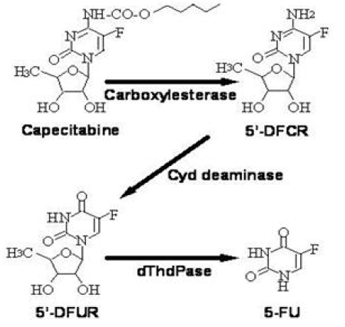
Enzymet dihydropyrimidindehydrogenase hydrogenerer 5-FU, produktet af capecitabin-metabolisme, til det meget mindre toksiske 5-fluor-5, 6-dihydro-fluorouracil (FUH2). Dihydropyrimidinase spalter pyrimidinringen for at give 5-fluor-ureido-propionsyre (FUPA). Endelig spalter β-ureido-propionase FUPA til α-fluor-β-alanin (FBAL), som renses i urinen.
In vitro enzymatiske undersøgelser med humane levermikrosomer indikerede, at capecitabin og dets metabolitter (5'-DFUR, 5'-DFCR, 5-FU og FBAL) ikke hæmmede metabolismen af testsubstrater af cytokrom P450 isoenzymer 1A2, 2A6, 3A4, 2C19, 2D6 og 2E1.
Udskillelse
Capecitabin og dets metabolitter udskilles overvejende i urinen; 95,5 % af den administrerede capecitabin-dosis genfindes i urinen. Fækal udskillelse er minimal (2,6%). Den vigtigste metabolit, der udskilles i urinen, er FBAL, som repræsenterer 57 % af den administrerede dosis. Ca. 3 % af den administrerede dosis udskilles i urinen som uændret lægemiddel. Eliminationshalveringstiden for både moder-capecitabin og 5-FU var ca. 0,75 time.
Effekt af alder, køn og race på farmakokinetikken af capecitabin
En populationsanalyse af poolede data fra de to store kontrollerede undersøgelser af patienter med metastatisk kolorektal cancer (n=505), som fik XELODA 500 mg ved 1250 mg/m2 to gange dagligt indikerede, at køn (202 kvinder og 303 mænd) og race (455 hvide/kaukasiske patienter, 22 sorte patienter og 28 patienter af anden race) har ingen indflydelse på farmakokinetikken af 5'DFUR, 5-FU og FBAL. Alder har ingen signifikant indflydelse på farmakokinetikken af 5'-DFUR og 5-FU i intervallet 27 til 86 år. En stigning på 20 % i alder resulterer i en stigning på 15 % i AUC af FBAL [se ADVARSLER OG FORHOLDSREGLER og DOSERING OG ADMINISTRATION ].
Efter oral administration af 825 mg/m2 capecitabin to gange dagligt i 14 dage havde japanske patienter (n=18) ca. 36 % lavere Cmax og 24 % lavere AUC for capecitabin end de kaukasiske patienter (n=22). Japanske patienter havde også omkring 25 % lavere Cmax og 34 % lavere AUC for FBAL end de kaukasiske patienter. Den kliniske betydning af disse forskelle er ukendt. Der forekom ingen signifikante forskelle i eksponeringen for andre metabolitter (5'-DFCR, 5'-DFUR og 5FU).
Virkning af leverinsufficiens
XELODA er blevet evalueret hos 13 patienter med mild til moderat leverdysfunktion på grund af levermetastaser defineret ved en sammensat score, herunder bilirubin, ASAT/ALT og alkalisk fosfatase efter en enkelt dosis på 1255 mg/m2 XELODA. Både AUC0-∞ og Cmax for capecitabin steg med 60 % hos patienter med leverdysfunktion sammenlignet med patienter med normal leverfunktion (n=14). AUC0-∞ og Cmax for 5-FU blev ikke påvirket. Hos patienter med mild til moderat leverdysfunktion på grund af levermetastaser skal der udvises forsigtighed, når XELODA 500 mg administreres. Virkningen af alvorlig leverdysfunktion på XELODA 500 mg er ikke kendt [se ADVARSLER OG FORHOLDSREGLER og Brug i særlige populationer ].
Virkning af nyreinsufficiens
Efter oral administration af 1250 mg/m2 capecitabin to gange dagligt til cancerpatienter med varierende grader af nedsat nyrefunktion, viste patienter med moderat (kreatininclearance = 30 til 50 ml/min) og svær (kreatininclearance 80 ml/min). Systemisk eksponering for 5'-DFUR var 42 % og 71 % større hos henholdsvis moderat og svært nedsat nyrefunktion end hos normale patienter. Systemisk eksponering for capecitabin var omkring 25 % større hos både moderat og svært nedsat nyrefunktion [se DOSERING OG ADMINISTRATION , KONTRAINDIKATIONER , ADVARSLER OG FORHOLDSREGLER , og Brug i særlige populationer ].
Effekt af capecitabin på farmakokinetikken af warfarin
Hos fire patienter med cancer øgede kronisk administration af capecitabin (1250 mg/m2 bid) med en enkelt 20 mg dosis warfarin den gennemsnitlige AUC for S-warfarin med 57 % og reducerede dets clearance med 37 %. Baseline korrigeret AUC for INR hos disse 4 patienter steg med 2,8 gange, og den maksimalt observerede gennemsnitlige INR værdi blev øget med 91 % [se KASSE ADVARSEL og DRUGSINTERAKTIONER ].
Effekt af antacida på farmakokinetikken af capecitabin
Da Maalox® (20 mL), et antacidum indeholdende aluminiumhydroxid og magnesiumhydroxid, blev administreret umiddelbart efter XELODA (1250 mg/m2, n=12 cancerpatienter), steg AUC og Cmax med henholdsvis 16 % og 35 %. for capecitabin og med henholdsvis 18% og 22% for 5'-DFCR. Der blev ikke observeret nogen effekt på de andre tre hovedmetabolitter (5'-DFUR, 5-FU, FBAL) af XELODA.
Effekt af Allopurinol på Capecitabin
Publiceret litteratur rapporterede, at samtidig brug med allopurinol kan reducere omdannelsen af capecitabin til de aktive metabolitter, FdUMP og FUTP; den kliniske betydning var dog ikke fuldt karakteriseret.
Effekt af Capecitabine på farmakokinetikken af docetaxel og vice versa
Et fase 1-studie evaluerede effekten af XELODA på farmakokinetikken af docetaxel (Taxotere®), og effekten af docetaxel på farmakokinetikken af XELODA blev udført hos 26 patienter med solide tumorer. XELODA viste sig ikke at have nogen effekt på farmakokinetikken af docetaxel (Cmax og AUC), og docetaxel har ingen effekt på farmakokinetikken af capecitabin og 5-FU precursoren 5'-DFUR.
Kliniske Studier
Adjuverende tyktarmskræft
Et multicenter randomiseret, kontrolleret fase 3 klinisk forsøg med patienter med Dukes' C colon cancer (X-ACT) gav data vedrørende brugen af XELODA til adjuverende behandling af patienter med colon cancer. Det primære formål med undersøgelsen var at sammenligne sygdomsfri overlevelse (DFS) hos patienter, der fik XELODA, med dem, der fik IV 5-FU/LV alene. I dette forsøg blev 1987 patienter randomiseret enten til behandling med XELODA 1250 mg/m2 oralt to gange dagligt i 2 uger efterfulgt af en 1-uges hvileperiode, givet som 3-ugers cyklusser i i alt 8 cyklusser (24 uger) eller IV. bolus 5-FU 425 mg/m2 og 20 mg/m2 IV leucovorin på dag 1 til 5, givet som 4-ugers cyklusser i i alt 6 cyklusser (24 uger). Patienterne i undersøgelsen skulle være mellem 18 og 75 år gamle med histologisk bekræftet Dukes' stadium C tyktarmskræft med mindst én positiv lymfeknude og have gennemgået (inden for 8 uger før randomisering) fuldstændig resektion af den primære tumor uden makroskopisk eller mikroskopisk tegn på resterende tumor. Patienterne skulle heller ikke have nogen forudgående cytotoksisk kemoterapi eller immunterapi (undtagen steroider) og have en ECOG-præstationsstatus på 0 eller 1 (KPS ≥ 70%), ANC ≥ 1,5x109/L, blodplader ≥ 100x109/L, serumkreatinin ≤ 1,5 ULN, total bilirubin ≤ 1,5 ULN, AST/ALT ≤ 2,5 ULN og CEA inden for normale grænser på tidspunktet for randomisering.
Baseline-demografien for XELODA- og 5-FU/LV-patienter er vist i tabel 10. Baseline-karakteristikaene var velafbalancerede mellem armene.
Alle patienter med normal nyrefunktion eller let nedsat nyrefunktion begyndte behandlingen med den fulde startdosis på 1250 mg/m2 oralt to gange dagligt. Startdosis blev reduceret hos patienter med moderat nedsat nyrefunktion (beregnet kreatininclearance 30 til 50 ml/min) ved baseline [se DOSERING OG ADMINISTRATION ]. Efterfølgende blev doser for alle patienter justeret efter behov i henhold til toksicitet. Dosisstyring for XELODA 500 mg inkluderede dosisreduktioner, cyklusforsinkelser og behandlingsafbrydelser (se tabel 11).
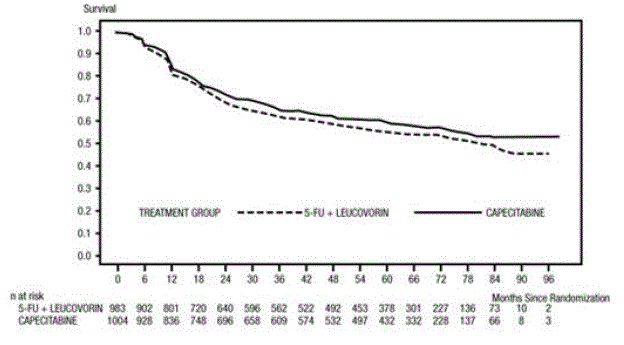
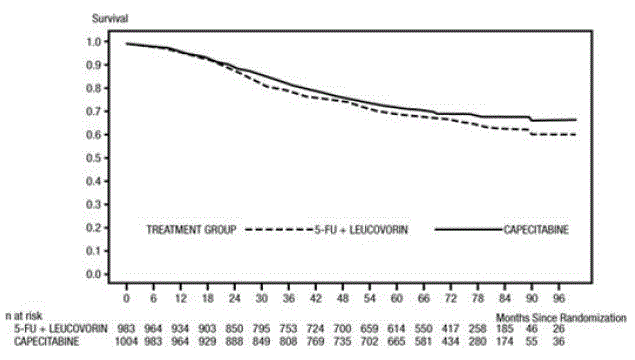
Medianopfølgningen på analysetidspunktet var 83 måneder (6,9 år). Hazard ratio for DFS for XELODA 500mg sammenlignet med 5-FU/LV var 0,88 (95 % CI 0,77 – 1,01) (se og figur 1 ). Fordi den øvre 2-sidede 95 % konfidensgrænse for hazard ratio var mindre end 1,20, var XELODA ikke ringere end 5-FU/LV. Valget af non-inferiority marginen på 1,20 svarer til tilbageholdelsen af cirka 75 % af 5-FU/LV-effekten på DFS. Hazard ratio for XELODA sammenlignet med 5-FU/LV med hensyn til samlet overlevelse var 0,86 (95 % CI 0,74 – 1,01). De 5-årige samlede overlevelsesrater var 71,4 % for XELODA og 68,4 % for 5-FU/LV (se figur 2).
Figur 1 Kaplan-Meier-estimater af sygdomsfri overlevelse (alle randomiserede populationer)a
Figur 2 Kaplan-Meier-estimater af samlet overlevelse (alle randomiserede populationer)
Metastatisk tyktarmskræft
Generel
Den anbefalede dosis af XELODA 500 mg blev bestemt i et åbent, randomiseret klinisk studie, hvor man udforskede effektiviteten og sikkerheden af kontinuerlig behandling med capecitabin (1331 mg/m2/dag i to opdelte doser, n=39), intermitterende behandling med capecitabin ( 2510 mg/m2/dag i to opdelte doser, n=34), og intermitterende behandling med capecitabin i kombination med oral leucovorin (LV) (capecitabin 1657 mg/m2/dag i to opdelte doser, n=35; leucovorin 60 mg/dag dag) hos patienter med fremskreden og/eller metastatisk kolorektalt karcinom i første-linje metastatisk indstilling. Der var ingen tilsyneladende fordel i responsraten ved at tilføje leucovorin til XELODA; dog var toksiciteten øget. XELODA, 1250 mg/m2 to gange dagligt i 14 dage efterfulgt af 1 uges hvile, blev udvalgt til yderligere klinisk udvikling baseret på den overordnede sikkerheds- og effektprofil for de tre undersøgte skemaer.
Monoterapi
Data fra to åbne, multicenter, randomiserede, kontrollerede kliniske forsøg med 1207 patienter understøtter brugen af XELODA i førstelinjebehandlingen af patienter med metastatisk kolorektalt karcinom. De to kliniske studier var identiske i design og blev udført i 120 centre i forskellige lande. Studie 1 blev udført i USA, Canada, Mexico og Brasilien; Undersøgelse 2 blev udført i Europa, Israel, Australien, New Zealand og Taiwan. I alt blev 603 patienter i begge forsøg randomiseret til behandling med XELODA 500 mg i en dosis på 1250 mg/m2 to gange dagligt i 2 uger efterfulgt af en 1-uges hvileperiode og givet som 3-ugers cyklusser; 604 patienter blev randomiseret til behandling med 5-FU og leucovorin (20 mg/m2 leucovorin IV efterfulgt af 425 mg/m2 IV bolus 5-FU, på dag 1 til 5, hver 28. dag).
begge forsøg blev den samlede overlevelse, tid til progression og responsrate (komplet plus delvis respons) vurderet. Svarene blev defineret af Verdenssundhedsorganisationens kriterier og forelagt en blindet uafhængig revisionskomité (IRC). Forskelle i vurderinger mellem investigator og IRC blev afstemt af sponsoren, blindet til behandlingsarmen, ifølge en specificeret algoritme. Overlevelse blev vurderet ud fra en non-inferioritetsanalyse.
Basislinjedemografien for XELODA- og 5-FU/LV-patienter er vist i tabel 13.
Effekt-endepunkterne for de to fase 3-forsøg er vist i og
Figur 3 Kaplan-Meier-kurve for samlet overlevelse af samlede data (undersøgelse 1 og 2)
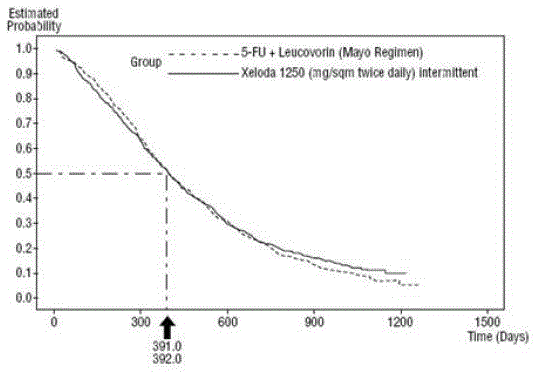
XELODA var bedre end 5-FU/LV for objektiv responsrate i undersøgelse 1 og undersøgelse 2. Ligheden mellem XELODA 500 mg og 5-FU/LV i disse undersøgelser blev vurderet ved at undersøge den potentielle forskel mellem de to behandlinger. For at sikre, at XELODA 500mg har en klinisk meningsfuld overlevelseseffekt, blev der udført statistiske analyser for at bestemme procentdelen af overlevelseseffekten af 5-FU/LV, som blev bibeholdt af XELODA. Estimatet af overlevelseseffekten af 5-FU/LV blev afledt af en meta-analyse af ti randomiserede undersøgelser fra den publicerede litteratur, der sammenlignede 5-FU med regimer af 5-FU/LV, der svarede til kontrolarmene anvendt i disse undersøgelser 1 og 2. Metoden til at sammenligne behandlingerne var at undersøge worst case (95 % konfidens øvre grænse) for forskellen mellem 5-FU/LV og XELODA 500 mg, og at vise, at tab på mere end 50 % af 5- FU/LV overlevelseseffekt blev udelukket. Det blev påvist, at procentdelen af den bibeholdte overlevelseseffekt af 5-FU/LV var mindst 61 % for undersøgelse 2 og 10 % for undersøgelse 1. Det samlede resultat stemmer overens med en retention på mindst 50 % af effekten af 5 -FU/LV. Det skal bemærkes, at disse værdier for bevaret effekt er baseret på den øvre grænse af 5-FU/LV vs XELODA 500 mg forskel. Disse resultater udelukker ikke muligheden for ægte ækvivalens mellem XELODA 500 mg og 5-FU/LV (se og Figur 3 ).
Brystkræft
XELODA er blevet evalueret i kliniske forsøg i kombination med docetaxel (Taxotere®) og som monoterapi.
I kombination med Docetaxel
Dosis af XELODA anvendt i det kliniske fase 3-studie i kombination med docetaxel var baseret på resultaterne af et fase 1-studie, hvor en række doser af docetaxel administreret i 3-ugers cyklusser i kombination med et intermitterende regime af XELODA (14 dage). behandling, efterfulgt af en 7-dages hvileperiode) blev evalueret. Kombinationsdosisregimet blev valgt baseret på tolerabilitetsprofilen for de 75 mg/m2 administreret i 3-ugers cyklusser af docetaxel i kombination med 1250 mg/m2 to gange dagligt i 14 dage med XELODA 500 mg administreret i 3-ugers cyklusser. Den godkendte dosis på 100 mg/m2 docetaxel administreret i 3-ugers cyklusser var kontrolarmen i fase 3-studiet.
XELODA i kombination med docetaxel blev vurderet i et åbent, multicenter, randomiseret forsøg i 75 centre i Europa, Nordamerika, Sydamerika, Asien og Australien. I alt 511 patienter med metastaserende brystkræft, der var resistente over for eller tilbagevendende under eller efter en antracyklinholdig behandling, eller recidiverende under eller recidiverende inden for 2 år efter at have afsluttet en antracyklinholdig adjuverende behandling. To hundrede og femoghalvtreds (255) patienter blev randomiseret til at modtage XELODA 1250 mg/m2 to gange dagligt i 14 dage efterfulgt af 1 uge uden behandling og docetaxel 75 mg/m2 som en 1-times intravenøs infusion administreret i 3-ugers cyklusser. I monoterapiarmen fik 256 patienter docetaxel 100 mg/m2 som en 1-times intravenøs infusion administreret i 3-ugers cyklusser. Patientdemografi er angivet i tabel 16.
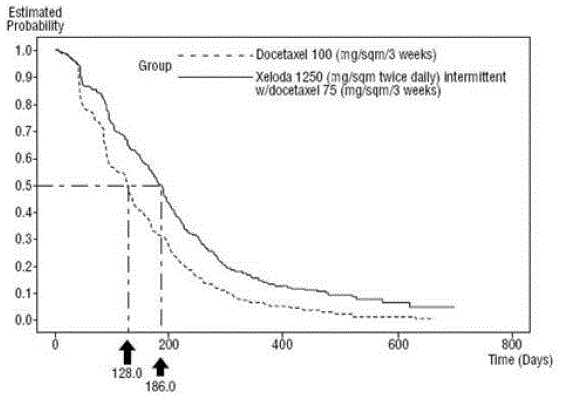
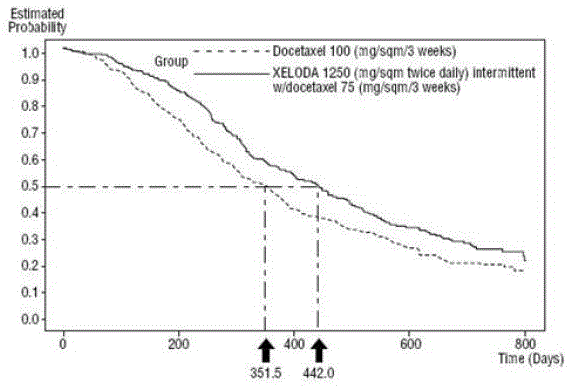
XELODA i kombination med docetaxel resulterede i statistisk signifikant forbedring i tid til sygdomsprogression, samlet overlevelse og objektiv responsrate sammenlignet med monoterapi med docetaxel som vist i og Figur 5 .
Figur 4 Kaplan-Meier-estimater for tid til sygdomsprogression XELODA og Docetaxel vs. Docetaxel
Figur 5 Kaplan-Meier estimater af overlevelse XELODA og Docetaxel vs. Docetaxel
Monoterapi
Antitumoraktiviteten af XELODA som monoterapi blev evalueret i et åbent enkeltarmsstudie udført i 24 centre i USA og Canada. I alt 162 patienter med stadium IV brystkræft blev indskrevet. Det primære endepunkt var tumorresponsrate hos patienter med målbar sygdom, med respons defineret som et ≥50 % fald i summen af produkterne af de vinkelrette diametre af bidimensionelt målbar sygdom i mindst 1 måned. XELODA blev administreret i en dosis på 1255 mg/m2 to gange dagligt i 2 uger efterfulgt af en 1-uges hvileperiode og givet som 3 ugers cyklusser. Baseline demografi og kliniske karakteristika for alle patienter (n=162) og dem med målbar sygdom (n=135) er vist i . Resistens blev defineret som progressiv sygdom under behandling, med eller uden et indledende respons, eller tilbagefald inden for 6 måneder efter afsluttet behandling med et antracyklinholdigt adjuverende kemoterapiregime.
Antitumorresponser for patienter med sygdomsresistente over for både paclitaxel og et antracyklin er vist i
For undergruppen på 43 patienter, som var dobbelt resistente, var mediantiden til progression 102 dage, og medianoverlevelsen var 255 dage. Den objektive responsrate i denne population blev understøttet af en responsrate på 18,5 % (1 CR, 24 PR'er) i den samlede population på 135 patienter med målbar sygdom, som var mindre resistente over for kemoterapi (se ). Den gennemsnitlige tid til progression var 90 dage, og den gennemsnitlige overlevelse var 306 dage.
PATIENTOPLYSNINGER
XELODA® (zeh-LOE-duh) (capecitabin) tabletter
Hvad er den vigtigste information, jeg bør vide om XELODA 500mg?
XELODA 500mg kan forårsage alvorlige bivirkninger, herunder:
- XELODA kan interagere med blodfortyndende medicin, såsom warfarin (COUMADIN®). Hvis du tager XELODA 500 mg sammen med disse lægemidler, kan det forårsage ændringer i, hvor hurtigt dine blodpropper og kan forårsage blødninger, der kan føre til døden. Dette kan ske så snart som et par dage efter, du er begyndt at tage XELODA 500 mg, eller senere under behandlingen, og muligvis endda inden for 1 måned efter, at du holder op med at tage XELODA. Din risiko kan være højere, fordi du har kræft, og hvis du er over 60 år.
- Inden du tager XELODA, skal du fortælle din læge, hvis du tager warfarin (COUMADIN) eller en anden blodfortyndende medicin.
- Hvis du tager warfarin (COUMADIN) eller et andet blodfortyndende middel, der ligner warfarin (COUMADIN) under behandling med XELODA, bør din læge ofte tage blodprøver for at kontrollere, hvor hurtigt dine blodpropper under og efter, du stopper behandlingen med XELODA. Din læge kan ændre din dosis af den blodfortyndende medicin, hvis det er nødvendigt.
Se "Hvad er de mulige bivirkninger af XELODA 500mg?" for mere information om bivirkninger.
Hvad er XELODA?
XELODA er en receptpligtig medicin, der bruges til at behandle mennesker med:
- kræft i tyktarmen, der har spredt sig til lymfeknuder i området tæt på tyktarmen (Dukes' C-stadie), efter de er blevet opereret.
- kræft i tyktarmen eller endetarmen (kolorektal), der har spredt sig til andre dele af kroppen (metastatisk), som din første behandling af din kræft på dette stadium.
- brystkræft, der har spredt sig til andre dele af kroppen (metastatisk) sammen med et andet lægemiddel kaldet docetaxel, efter at behandling med visse andre lægemidler mod kræft ikke har virket.
- brystkræft, der spredes til andre dele af kroppen og ikke er blevet bedre efter behandling med paclitaxel og visse andre kræftlægemidler, eller som ikke kan modtage mere behandling med visse kræftlægemidler.
Det vides ikke, om XELODA 500mg er sikkert og effektivt til børn.
Tag ikke XELODA, hvis du:
- har alvorlige nyreproblemer
- er allergisk over for capecitabin, 5-fluorouracil eller nogen af dets ingredienser i XELODA. Se slutningen af denne indlægsseddel for en komplet liste over ingredienser i XELODA.
Tal med din læge, før du tager XELODA, hvis du ikke er sikker på, om du har nogen af ovenstående tilstande.
Inden du tager XELODA, skal du fortælle din læge om alle dine medicinske tilstande, inklusive hvis du:
Se "Hvad er den vigtigste information, jeg bør vide om XELODA?"
- har haft hjerteproblemer
- har nyre- eller leverproblemer
- har fået at vide, at du mangler enzymet DPD (dihydropyrimidin dehydrogenase).
- er gravid eller planlægger at blive gravid. XELODA 500mg kan skade dit ufødte barn. Din læge bør lave en graviditetstest, før du starter behandling med XELODA. Fortæl din læge med det samme, hvis du bliver gravid eller tror, du kan være gravid under behandling med XELODA.
- Kvinder som er i stand til at blive gravide, skal bruge effektiv prævention under behandlingen og i 6 måneder efter den sidste dosis. Tal med din læge om præventionsvalg, der kan være rigtige for dig under behandling med XELODA.
- Hanner som har kvindelige partnere, der er i stand til at blive gravide, bør bruge effektiv prævention under behandlingen og i 3 måneder efter den sidste dosis.
- ammer eller planlægger at amme. Det vides ikke, om XELODA udskilles i din modermælk. Du må ikke amme under behandling med XELODA 500 mg og i 2 uger efter den sidste dosis.
Fortæl din sundhedsudbyder om al den medicin, du tager, inklusive receptpligtig og håndkøbsmedicin, vitaminer og naturlægemidler. XELODA kan påvirke den måde, anden medicin virker på, og anden medicin kan påvirke den måde, XELODA virker på.
Kend den medicin du tager. Hold en liste over dem for at vise din læge og apotek, når du får en ny medicin.
Hvordan skal jeg tage XELODA 500mg?
- Tag XELODA nøjagtigt som din læge fortæller dig at tage det.
- Din læge vil fortælle dig, hvor meget XELODA du skal tage, og hvornår du skal tage det.
- Tag XELODA 2 gange om dagen, 1 gang om morgenen og 1 gang om aftenen.
- Tag XELODA 500 mg inden for 30 minutter efter at have afsluttet et måltid.
- Synk XELODA 500 mg tabletter hele med vand. Lade være med knuse eller skære XELODA 500 mg tabletter. Hvis du ikke kan sluge XELODA-tabletter hele, skal du fortælle det til din læge.
- Din læge kan ændre din dosis, stoppe midlertidigt eller permanent stoppe behandlingen med XELODA 500 mg, hvis du udvikler bivirkninger.
- Hvis du tager for meget XELODA, skal du straks ringe til din læge eller gå til den nærmeste skadestue.
Hvad er de mulige bivirkninger af XELODA?
XELODA kan forårsage alvorlige bivirkninger, herunder:
Kvalme og opkastning er almindelige med XELODA. Hvis du mister appetitten, føler dig svag og får kvalme, opkastning eller diarré, kan du hurtigt blive dehydreret.
Stop med at tage XELODA og ring med det samme til din læge, hvis du:
Hvis dit antal hvide blodlegemer er meget lavt, har du øget risiko for infektion. Ring til din læge med det samme, hvis du får feber på 100,5 oF eller mere eller har andre tegn og symptomer på infektion.
- Se "Hvad er den vigtigste information, jeg bør vide om XELODA?"
- Diarré. Diarré er almindelig med XELODA og kan nogle gange være alvorlig. Stop med at tage XELODA og ring med det samme til din læge, hvis antallet af afføringer, du har på en dag, stiger med 4 eller flere afføringer, end det er normalt for dig, eller afføring om natten. Spørg din læge om, hvilken medicin du kan tage for at behandle din diarré. Hvis du har svær blodig diarré med svære mavesmerter og feber, skal du stoppe med at tage Xeloda 500 mg og ringe til din læge eller gå til den nærmeste skadestue.
- Hjerteproblemer. XELODA kan forårsage hjerteproblemer, herunder: hjerteanfald og nedsat blodgennemstrømning til hjertet, brystsmerter, uregelmæssige hjerteslag, ændringer i dit hjertes elektriske aktivitet set på et elektrokardiogram (EKG), problemer med din hjertemuskel, hjertesvigt og pludselige død. Stop med at tage XELODA 500mg og ring til din læge eller gå til den nærmeste skadestue, hvis du får nye symptomer på et hjerteproblem, herunder:
- brystsmerter
- svimmelhed
- stakåndet
- svimmelhed
- Tab af for meget kropsvæske (dehydrering) og nyresvigt. Dehydrering kan ske med XELODA 500mg og kan forårsage pludselig nyresvigt, der kan føre til døden. Du er i højere risiko, hvis du har nyreproblemer, før du tager XELODA 500 mg og også tager anden medicin, der kan forårsage nyreproblemer.
- kaste op 2 eller flere gange på en dag.
- kun kan spise eller drikke lidt nu og da, eller slet ikke på grund af kvalme.
- har diarré. Se "diarré" ovenfor.
- Alvorlige hud- og mundreaktioner.
- XELODA 500mg kan forårsage alvorlige hudreaktioner, der kan føre til døden. Fortæl din læge med det samme, hvis du udvikler hududslæt, blærer og afskalning af din hud. Din læge kan fortælle dig, at du skal stoppe med at tage XELODA, hvis du får en alvorlig hudreaktion. Tag ikke XELODA 500mg igen, hvis dette sker.
- XELODA 500mg kan også forårsage "hånd og fod"-syndrom. Hånd- og fodsyndrom er almindeligt med XELODA 500 mg og kan forårsage følelsesløshed og ændringer i følelsen i hænder og fødder eller forårsage rødme, smerte, hævelse af hænder og fødder. Stop med at tage XELODA og ring med det samme til din læge, hvis du har nogle af disse symptomer, og du ikke er i stand til at udføre dine sædvanlige aktiviteter.
- Hånd- og fodsyndrom kan føre til tab af fingeraftryk, hvilket kan påvirke din identifikation.
- du kan få sår i munden eller på tungen, når du tager XELODA. Stop med at tage XELODA 500mg og ring til din læge, hvis du får smertefuld rødme, hævelse eller sår i munden eller tungen, eller hvis du har problemer med at spise.
- Øget niveau af bilirubin i dit blod og leverproblemer. Øget bilirubin i dit blod er almindeligt med XELODA 500 mg og kan også nogle gange være alvorligt. Din læge vil tjekke dig for disse problemer under behandling med XELODA.
- Nedsat antal hvide blodlegemer, blodplader og røde blodlegemer. Din læge vil tage blodprøver under behandling med XELODA for at kontrollere dit blodcelletal.
Personer på 80 år eller ældre kan være mere tilbøjelige til at udvikle alvorlige eller alvorlige bivirkninger med XELODA.
De mest almindelige bivirkninger af XELODA 500mg omfatter:
- diarré
- smerter i maveområdet (mave).
- hånd- og fodsyndrom
- svaghed og træthed
- kvalme
- brækkede sig
- øgede mængder af røde blodlegemers nedbrydningsprodukter (bilirubin i dit blod
Alvorlige allergiske reaktioner kan forekomme med XELODA. Fortæl din læge, hvis du nogensinde har haft en allergisk reaktion over for capecitabin eller 5-fluorouracil. Se "Tag ikke XELODA, hvis du:". Stop med at tage XELODA 500mg og ring med det samme til din læge eller gå til en skadestue, hvis du har nogle af følgende symptomer på en alvorlig allergisk reaktion:
- røde kløende pletter på din hud (nældefeber)
- hud rødme
- hævelse af dit ansigt, læber, tunge eller hals
- udslæt
- kløe
- problemer med at synke eller trække vejret
XELODA 500mg kan forårsage fertilitetsproblemer hos kvinder og mænd. Dette kan påvirke evnen til at få et barn. Tal med din læge, hvis du har bekymringer om fertilitet.
Disse er ikke alle de mulige bivirkninger af XELODA.
Ring til din læge for lægehjælp om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
Hvordan skal jeg opbevare XELODA?
- Opbevar XELODA 500mg ved stuetemperatur mellem 68°F til 77°F (20°C til 25°C).
- Opbevar XELODA 500mg i en tæt lukket beholder.
- Spørg din læge eller apotekspersonalet, hvordan du sikkert smider ubrugt XELODA ud.
Opbevar XELODA og al medicin utilgængeligt for børn.
Generel information om sikker og effektiv brug af XELODA.
Lægemidler ordineres nogle gange til andre formål end dem, der er anført i en patientinformationsfolder. Brug ikke XELODA til en tilstand, som den ikke er ordineret til. Giv ikke XELODA 500 mg til andre mennesker, selvom de har de samme symptomer, som du har. Det kan skade dem. Du kan spørge din apoteker eller sundhedsudbyder om oplysninger om XELODA 500mg, der er skrevet til sundhedspersonale.
Hvad er ingredienserne i XELODA 500mg?
Aktiv ingrediens: capecitabin
Inaktive ingredienser: vandfri lactose, croscarmellosenatrium, hydroxypropylmethylcellulose, mikrokrystallinsk cellulose, magnesiumstearat og renset vand. Belægningen af fersken eller lys ferskenfilm indeholder hydroxypropylmethylcellulose, talkum, titaniumdioxid og syntetiske gule og røde jernoxider.
For mere information, gå til http://www.gene.com/patients/medicines/xeloda 500mg eller ring på 1-877-436-3683.
Denne patientinformation er blevet godkendt af US Food and Drug Administration.